

Volume 2, Issue 3 (August 2023)
Health Science Monitor 2023, 2(3): 180-204 |
Back to browse issues page


Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Tourani M, Samavarchi Tehrani S, Movahedpour A, Rezaei Arablouydareh S, Maleksabet A, Savardashtaki A, et al . Design and evaluation of a multi-epitope vaccine for COVID-19: an in silico approach. Health Science Monitor 2023; 2 (3) :180-204
URL: http://hsm.umsu.ac.ir/article-1-119-en.html
URL: http://hsm.umsu.ac.ir/article-1-119-en.html
Mehdi Tourani * 

 , Sadra Samavarchi Tehrani , Ahmad Movahedpour , Sahar Rezaei Arablouydareh , Amir Maleksabet , Amir Savardashtaki , Hojat Ghasemnejad‐Berenji , Mortaza Taheri-Anganeh
, Sadra Samavarchi Tehrani , Ahmad Movahedpour , Sahar Rezaei Arablouydareh , Amir Maleksabet , Amir Savardashtaki , Hojat Ghasemnejad‐Berenji , Mortaza Taheri-Anganeh
, Sadra Samavarchi Tehrani , Ahmad Movahedpour , Sahar Rezaei Arablouydareh , Amir Maleksabet , Amir Savardashtaki , Hojat Ghasemnejad‐Berenji , Mortaza Taheri-Anganeh
Cellular and Molecular Biology Research Center, Health Research Institute, Babol University of Medical Sciences, Babol, Iran
Full-Text [PDF 1157 kb]
(1742 Downloads)
| Abstract (HTML) (2941 Views)
Table 1. Consensus linear B cell epitopes predicted by ABCpred and BepiPred-2.0
Table 2. High scored and consensus cytotoxic T cells (CTL) epitopes predicted by NetCTL and CTLPred
Table 3. Highest scored helper T cells (HTL) epitopes predicted by NetMHCII 2.2.
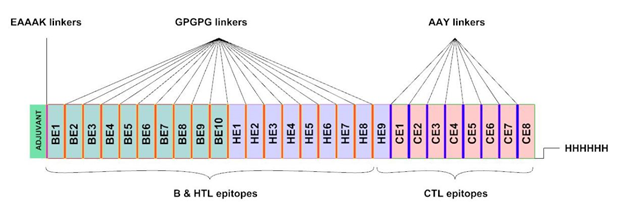
Fig 1. The multi-epitope vaccine construct. The adjuvant resides on N-terminus (green). BEs, HEs, and CE represent B cell linear epitopes, helper T cell epitopes, and cytotoxic T cell epitopes, respectively. The His-tag for affinity purification is located on C-terminus.
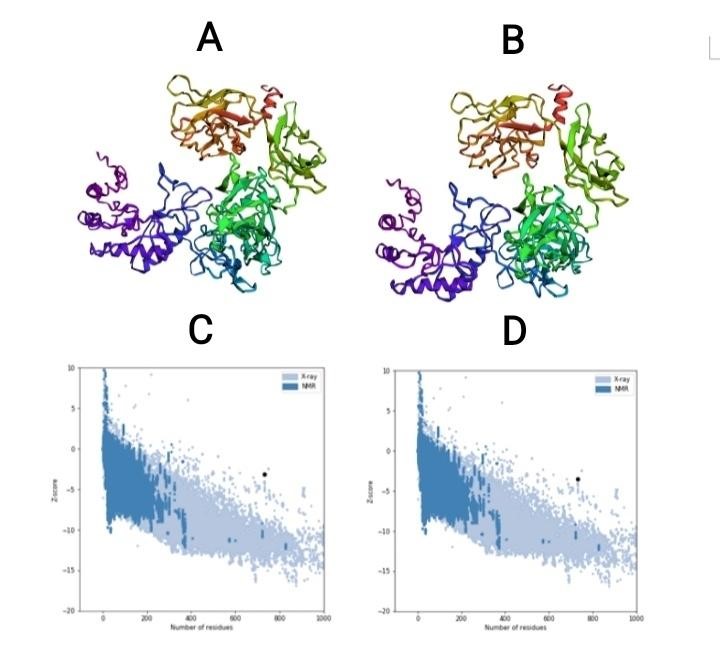
Fig 2. (A) 3D model before refinement. (B) 3D model after refinement. (C) Z-score before 3D model refinement. (D) Z-score after 3D model refinement.
Table 4. Comparison of residues residence in Ramachandran plot before and after model refinement.
Codon optimization:
The coding sequence of the recombinant vaccine was optimized for expression in E. coli using GenScript’s Multi-parameter Gene Optimization algorithm, OptimumGeneTM (http://www.genescript.com/). The codon optimized sequence had a length of 2205 nucleotides. The codon adaptation index (CAI), the frequency of optimal codons (FOC), and the GC contents of the coding sequence were respectively 0.95, 84 %, and 61% following optimization, indicating the possibility of overexpression of the multi-epitope vaccine in the E. coli host. The suitable GC content is between 30% and 70%. Optimum over-expression of inserted sequence is more than 0.8 according to CAI. Un-adapted codons can be resulted in a minor expression rate. CAI score was also confirmed by JCat. Details of adapted codons are shown in figure 6B. Finally, the coding sequence of designed vaccine was inserted into pET28a (+) and the recombinant plasmid was designed by VectorNTI (Figure 9).
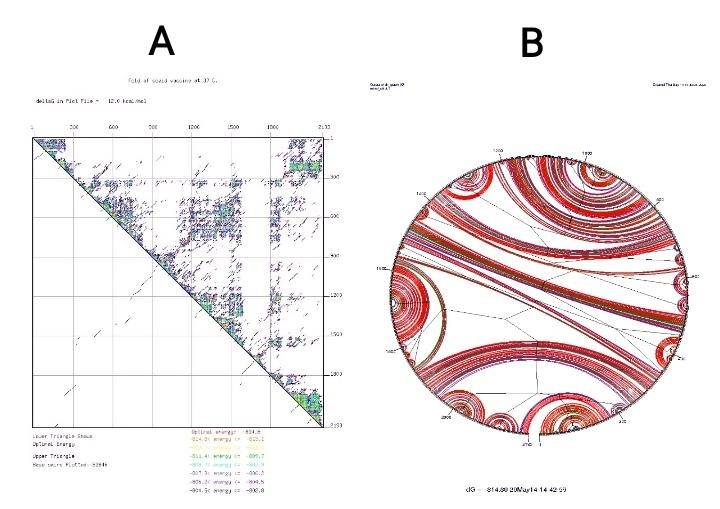
Fig. 8. (A) Point energy diagram for mRNA structure. (B) Circular energy diagram for mRNA structure.
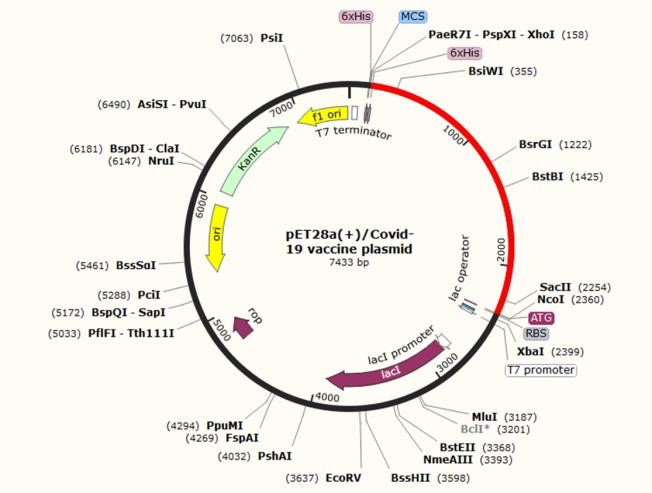
Fig. 9. In silico cloning of the final vaccine construct into pET28a (+) expression vector, where the red part indicates the coding gene for the vaccine surrounded between XhoI (158) and NcoI (2360) while the vector backbone is shown in a black circle.
Discussion
While more than 100 years have passed since the first outbreak of influenza (flu) in 1918, we have faced the emergence of a novel pandemy, causing severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) at the beginning of 2020 (66). What sets this pandemic apart from others is the ability to transmit, which has spread to all continents and affected about 5 million people worldwide in a five-month period (67). The presence of mutations and antigen variability in the coronavirus structure has made it challenging to design and produce a specific vaccine (68).
In the present study, we designed a new chimeric protein vaccine consisting of 27 immunodominant epitopes from the spike protein of SARS-CoV-2. Spike glycoprotein, as a surface antigen with a crown-like appearance, is responsible for the initial binding of the coronavirus to lung cells. Therefore, it plays a pivotal role in entering the host cells (69). SARS-COV-2 spike protein has been shown to be phylogenetically close to bat coronavirus and binds with high affinity to ACE2-derived protein of human origin (70). Studies have shown that mice vaccinated with SARS-like coronaviruses S (SL-CoV) recombinant protein (which are structurally very similar to spike protein of SARS‐CoV‐2) have mounted strong humoral and cellular immune responses by producing high levels of IFN-γ and IL-6. Therefore, spike protein can be considered a suitable target to design vaccines against SARS-CoV-2 (71, 72).
Before the appearance of COVID-19, in 2008, the spike protein-related DNA vaccine was also used to evaluate anti-SARS immunity. In the study by Julie E et al., the vaccine was well tolerated. ELISA detected SARS-CoV-specific antibodies in 80% of cases, and neutralizing antibodies were detected in all individuals. SARS-CoV-specific T4 CD4 + SAR cellular responses were recognized in all cases and T + CD8 cell responses in 20% of individuals (73). Taken together, many vaccine platforms for SARS-CoV have employed spike protein in vaccine structures for immunization, including live attenuated recombinant virus, recombinant modified vaccinia Ankara (MVA) virus, recombinant non-replicating adenovirus (E-deleted), DNA-based vaccines, soluble proteins/adjuvant, virus-like particles (VLPs)/adjuvant, and a combination of vaccine approaches (DNA/peptides, DNA/recombinant viral vector, viral vector/peptides) (74).
In order to boost the immunogenicity of the chimeric vaccine, adjuvant was utilized. Meanwhile, the adjuvant joined with a vaccine intensifies its immunogenic potential and reduces the optimum dose. By mixing with a soluble and weak immunogen, an adjuvant can turn it into an insoluble and stronger immunogen because it retains its high concentration for a longer period of time using antigen depot. In fact, adjuvants are non-specific stimuli for dendritic cells-related to draining as well as cellular and humoral immune responses (75). Recently, TLR agonists have been used as powerful natural adjuvants in vaccine production. Adjuvants enhance the production of inflammatory cytokines, resulting in recruitment of cells of innate and adaptive immunity. In addition to the properties, it strengthens the intracellular cascades that produce inflammatory cytokines and further recruitment of the innate and adaptive immune system cells by stimulate the TLRs of antigen-presenting cells (APC) (76). In this study, the 50S ribosomal protein L7/L12 of Mycobacterium tuberculosis was utilized as an adjuvant and TLR4 agonist. by interacting with TLR4, the 50s ribosomal L7/L12 stimulates the proliferation and activation of immature dendritic and naïve T cells (58, 77).
Accordingly, we selected our favorite sequences considering T and B cell stimulation, high-affinity MHC Class I, MHC Class II, and linear B-cell epitopes from SARS‐CoV‐2 spike protein along with 50s ribosomal L7 / L12. The selected sequences were joined together via appropriate linkers to produce a stable multi-epitope peptide construct. Linkers play a key role in the area of recombinant fusion protein technology such as constructing multi-epitope vaccines. They help the proper folding of each functional domain within the structure of the multi-domain protein, increase the yield of expression in the host systems, and enhance the bioactivity of the recombinant fusion protein (78). In the present study, three types of linkers were used for vaccine construction. The AAY and GPGPG between the epitopes give the structure the ability to minimize junctional immunogenicity (79). AAY is a cleavage site of proteasomes thereby improving the epitope presentation by APCs to immune cells (80). Besides, GPGPG can facilitate the immune processing and epitope presentation by HLA-II to helper T lymphocytes, and is an effective stimulus for cellular immunity, as well (81). EAAAK linker was also used to bind viral antigens to the adjuvants. This linker improves the biological activity of epitopes and intensifies the expression of the final product in the bacterial host, creating a coherent structure (82). The absence of the linkers between the epitopes leads to the production of connective epitopes (neo-epitopes) and protein function disturbance (83).
Therefore, our multi-subunit vaccine sequence was selected with the 1029 length residues. The vaccine had interferon-γ (IFN γ) epitopes. The importance of IFN γ epitopes analysis is due to the fact that IFN-γ produced by CD4+ T cells improve neutrophils and macrophages recruitment using CXC family chemokines and functionally intensify the HTLs and CTLs (84). The presence of 149 positive IFN-γ inducing peptides within the structure of the multi-epitope vaccine implies that the vaccine can enhance the responses of cellular immunity vaccines carrying multiple epitopes which often have poor immunogenicity and require binding to amplifier compounds (85).
The antigenicity score was 0.4606 (close to threshold: 0.4) for the S protein-based vaccine. However, this score increased to 0.835593 when the sequence of the adjuvant was added to the vaccine. When designing a vaccine, it is also crucial to prevent or at least minimize the allergic reactions i.e. the vaccine should prevent the shift in Th1/Th2 balance towards Th2-type response (86). The molecular weight of the vaccine candidate was 76863.88 Da, so overexpressed recombinant protein solubility is expected to be acceptable in E. coli host. The isoelectric pH and the net charge of the protein were 8.66 and +7, respectively, indicating the slightest basic molecule in nature. The instability index was calculated 20.46, indicating that the vaccine is stable upon expression. Moreover, the aliphatic index was 76.18 indicating that there are appropriate aliphatic side chains which make the protein potentially thermostable. Furthermore, grand average of hydropathicity (GRAVY) of -0.164 represents hydrophilicity and efficient interaction with water molecules for better vaccine solubility (87). The solubility of the designed vaccine was also predicted by the SOLpro server.
Understanding the conformational structure of a target protein and their receptors is valuable, where it is possible to determine protein-protein interactions stability and eliminate structural disorders (88). Five 3D structures were predicted for the designed multi-epitope vaccine among which one model with high accuracy and high C-score (-1.15) was selected, and the structural similarities of the vaccine with native protein structures was investigated by considering the TM-score and RMSD. TM-score of 0.57 ± 0.15 and an RMSD of 10.9 ± 4.6% showed that the construct had a correct topology. The selected model was then refined 27% using Galaxy Refine server.
The Ramachandran plot showed whether the residues were found in the favored and allowed regions to confirm the refinement. Comparing the phi and psi dihedral angles of amino acid residues, Ramachandran plot disclosed that there are very few residues in the outlier and unfavorable regions. The ProSA-web server also showed that the original model has been modified (Z-Score=-3.15 to -3.55) after refinement and the designed structure is functionally approached by native proteins. Evaluation of the final three-dimensional structure in terms of discontinuous B-cell epitopes, from a practical point of view showed that epitopes 4 to 115 in length residues could interestingly stimulate antibody production. Immunogenic epitopes exposed on the vaccine structure can effectively induce the differentiation of the B cells to memory and plasma cells, resulting in more persistent protection against SARS-CoV-2 (89). Choudhury et al. reported that TLR4 plays a central role in identification of the molecular patterns of SARS-CoV-2 to generating inflammatory responses. TLR4 can interact with SARS-CoV-2 spike glycoprotein with a higher affinity than TLR1 and TLR6 (90). In vivo studies have also shown that TLR4 knockout mice have either similar survival rates in some cases or even more severe infection than wild-type mice infected with dengue virus (DENV), Ebola virus (EBOV), SARS-CoV, or respiratory syncytial virus (RSV) (91-94). Overall, it was found that virus infection-associated immune response may require TLR4 signaling activation (95).
Therefore, investigating the potential binding strength of the vaccine to TLR4 can be considered as a critical factor in evaluating its efficacy. Hence, we selected the best possible interaction model considering the docking process between the designed construct and TLR4, and evaluated the stability of the complex using elastic-mode analysis. The results showed that the TLR4: vaccine complex had admissible stability, so there is enough time to convey downstream signaling pathways.
The flexibility, compactness, and stability of purposed vaccine structure were also estimated using NAMD and VMD. The complex had a tendency to stabilize after about two nanoseconds and attained a more compact and stable structure during the MD simulation time. This structure may become more compact if the simulation time are increased. Overall picture of the protein residue fluctuation showed the existence of highly flexible areas and the strength of intermolecular interactions with VMD improved the number of hydrogen bonds, resulting in a more stability.
Rigid body docking, clustering of lowest energy structure, and structural refinement were consecutively conducted using energy minimization. The complex with the lowest energy was selected by the server to minimize the potential energy and to obtain stabilized docked complex. Replacing the appropriate protein atoms, inappropriate conformational structure was stabled using energy minimization. Moreover, construct motion stiffness was evaluated by eigenvalue to refine complex deformability. The relative stiffness was appropriately obtained as a functional 3D protein structure. The flexibility of a functional protein structure must be such that its conformational epitopes do not alter in different presumed physiological conditions. Ultimately, the production potential must be evaluated in a bacterial expression host to investigate the effectiveness of a designed vaccine. From a practical point of view, the gene sequence of the vaccine of the interest must be stable enough to be translated following the transcription process (96). Hairpin and pseudoknot at the 5' end of the secondary structure of mRNA can adversely affect the efficiency of the protein translation. The sequence length, the number and situation of loops, the GC content, and etc. must be evaluated before the in-vitro phase. Examination of these important indicators showed the stable status of the mRNA structure (97). The protein can be purified to assess the quality of its binding to the antibodies present in the serum of SARS- CoV-19 infected patient using serological tests. Therefore, codons were optimized in order to over-express the vaccine protein in a suitable host (E. coli strain K12). CAI, FOC, and GC Content were reported 0.95, 84%, and 61%, respectively, demonstrating the desirable overexpression potential. Changes in mentioned parameters can also have profound effects on gene expression. Based on the finding, it was predictable that inserting the vaccine sequence in the vector pET28a (+) would have an acceptable expression in E. coli.
Conclusion
Nowadays, a worldwide effort is being made to prevent and treat emerging viral infections. Meanwhile, predicting the efficiency of the vaccines before their production has become the main approach of all studies. That is why in silico studies have become an intelligent strategy for producing vaccines. Accordingly, in the present study, a multi-subunit vaccine was designed against SARS-CoV-2 using the antigen minimization approach. The vaccine was designed based on the most conserve and immunogenic epitopes of the SARS-CoV-2 spike protein, instead of focusing on the whole virus. The scores obtained from the immunoinformatics databases indicated that the construct could be considered an appropriate vaccine candidate to protect against COVID-19 infection. Indeed, in vitro and in vivo studies need to be performed to evaluate the production of the vaccine in prokaryotic host systems, as well as, investigating its efficacy in eliciting the immune responses in animal models.
Acknowledgments
None declared.
Conflict of interest
The authors have no conflict of interest in this study.
Funding/support
This study was financially supported by the office of vice-chancellor for research of Shiraz University of Medical Sciences.
Data availability
The raw data supporting the conclusions of this article are available from the authors upon reasonable request.
Full-Text: (1886 Views)
Introduction
Coronaviruses (CoVs) are a member of the subfamily Othocoronavirinae, in the family Coronaviridae (1-3). They are extremely pathogenic and are often known to be infectious, as shown by the 2002 and 2012 epidemic of severe acute respiratory syndrome (SARS) and middle east respiratory syndrome (MERS) (4, 5). In late 2019, an outbreak of another coronavirus that triggers a respiratory-related disease was identified in Wuhan, Hubei, China, a disease now publicly named “the Corona Virus Disease 2019; COVID-19” (6). Hence, World Health Organization (WHO) announced global health disaster and regarded the COVID-19 as pandemic (7, 8). The major clinical symptoms in patients with SARS-CoV-2 are fatigue, cough, trouble breathing, shortness of breath, increased levels of lactate dehydrogenase (LDH), and extended prothrombin period. The less common signs involved sputum development, headache, hemoptysis, and diarrhea (9, 10). Genome-wide phylogenetic study reveals that SARS-CoV-2 shares 79.5%, 50%, and 96% sequence similarity with SARS-CoV, MERS-CoV, and bat coronavirus, respectively (11). Unlike other coronaviruses, the new SARS-CoV-2 is a positive-sense single-stranded RNA virus. Moreover, its genome size ranges between about 26,000 and 32,000 bases, comprises a variable number of open reading frames (ORFs). This novel virus is spherical with a diameter of approximately 125 nm, as well as it encodes 9860 amino acids translating into many non-structural proteins such as replicas (orf1a / b), nsp2, nsp3 and accessory proteins (orf3a, orf7a / b) along with structural proteins including spike (S), envelope (E), membrane (M), and nucleocapsid (N) proteins (12-14). Of these structural proteins, the spike glycoprotein was shown to attach to the cellular receptor angiotensin-converting enzyme 2 (ACE2), facilitating the virus entry to the host cells (15-18). Structurally, the S protein belongs to the class I viral protein, which contains two functional subunits, an amino-terminal S1 (685 aa) and a carboxyl-terminal S2 (588 aa) subunit (19). Each of these two subunits has a specific task. The subunit S1 plays a critical role in the virus binding to the host cell receptors, while the subunit S2 is involved in virus-host membrane fusion. It was found that the S1 protein of SARS-CoV-2 shows approximately 70% similarity with that of human SARS-CoVs. This subunit has two main domains: I) an N-terminal domain (NTD) that mediates sugar binding, II) a C-terminal domain (CTD) that facilitates protein receptor recognition. Therefore, both domains are considered as receptor-binding domains (RBDs) (20-22). Numerous data indicate that the S protein, as a virus key antigen, plays a crucial role in stimulating the production of neutralizing antibodies which accordingly inhibit the virus entry to the host cells. Hence, blocking the initial entry of a virus is suggested as a valuable approach in managing various viral diseases (21, 23, 24).
Currently, there are several nonspecific approaches for the control of SARS-CoV-2, including administration of type I IFNs, ribavirin, lopinavir/ritonavir, remdesivir, nelfinavir, arbidol, Chloroquine, and convalescent plasma (25, 26). Although vaccination is regarded the most efficient modality in prevention of infection diseases, yet there is no approved specific vaccine for SARS-CoV-2 (25). (27-29).
Today, bioinformatics and computational biology have influenced various aspects of biology and medicine (30-32). Using computational approaches prior to beginning an experimental study, minimizes the costs and time while improving the precision and validity of the experimental studies (33-35). In recent years, in silico approaches have enabled us to design epitope-based vaccine against various diseases such as infectious diseases and cancers. These approaches speed up the process of designing, developing, and evaluation of vaccine candidates (32, 36-38). Multi-epitope vaccines are constructed utilizing the most immunodominant regions (components) of the pathogen's proteins which have conserved sequences that allows them more success and can support in opposing the high mutation frequency in RNA viruses. These types of vaccines have numerous advantages such as safety, efficiency, simple production in large-scale, cost-curtailing, antigenicity, and immunogenicity (39-42). An optimal vaccine should include both B-cell and T-cell epitopes, for which the vaccine may effectively either induce particular humoral and cellular immunity against pathogens (43). The production of antibodies by B cells activation is essential for maintaining immunity to coronavirus, along with the virus-specific killer and memory CD8 + T cells to eradicate infected cells. Given the contribution of multi-epitope vaccines in responses restricted via a broad range of HLA molecules and make a balanced CD4+ and CD8+ cellular immune response, this strategy can be useful in clinic (44, 45).
We decided to design a multi-epitope vaccine against SARS-CoV-2 based on the virus S protein, and evaluated its biochemical and immune stimulatory features using in silico approaches.
.
Materials & Methods
Antigen selection:
Recently, the genome of SARS-CoV-2 was sequenced and the proteins coded by the virus were identified. The amino acid sequence of spike (S) protein was picked up from NCBI Protein database under reference number: YP_009724390.1. This protein is 1273 amino acids long.
Prediction of linear B cell epitopes:
The B-cell epitopes of antigens are recognized by humoral immune system. This type of epitope is the specific part of the antigen which is a target for B cell receptors. These epitopes have an essential role in designing an efficient vaccine. Linear B-cell epitopes were evaluated mostly by the BepiPred-2.0 web server (http://www.cbs.dtu.dk/services/BepiPred/) and ABCpred prediction server (http://crdd.osdd.net/raghava/abcpred/). Since BepiPred-2.0 is a strict server, the threshold was considered 0.51. BepiPred-2.0 predicts according to a random forest algorithm which is expert for identifying epitopes from interactions of antibody-antigen. 14-mers epitopes were anticipated by ABCpred with a threshold of 0.85 because this server is more lenient. ABCpred predicts epitopes based on recurrent neural network method (46, 47).
Prediction of cytotoxic T lymphocytes (CTL) epitopes:
Cytotoxic T lymphocyte (CTL) epitopes of S antigen were evaluated via NetCTL 1.2 server (http://www.cbs.dtu.dk/services/NetCTL/). This server method contains combination of MHC I binding peptides, cleavage of C-terminus in proteasomes, and TAP (Transporter Associated with Antigen Processing) transport efficacy predictions. However, the prediction of CTL epitopes in this server is limited to 12-mers peptides for MHC I supertypes. In this study, only the HLA-A2 was chosen. The prediction of MHC I binding and peptide cleavage in proteasomes was carried out by artificial neural networks. TAP transportation was evaluated by a weight matrix (48). The predictions were sorted based on combined score. The threshold for the prediction of CTL epitopes was set at 0.75 (default). The sequence of the S antigen was submitted to CTLPred server (http://crdd.osdd.net/raghava/ctlpred/). The prediction method of CTLPred is depended on a sophisticated machine learning techniques called artificial neural network (ANN) and support vector machine (SVM) (49). The combined prediction method was selected for this study. The thresholds were at default scores.
Helper T-lymphocyte (HTL) epitopes prediction:
Prediction of 15-mers HTL epitopes for human alleles was carried out using NetMHCII 2.3 Server (http://www.cbs.dtu.dk/services/NetMHCII/). Epitope peptides that bind HLA-DR, HLA-DQ, and HLA-DP alleles of human were chosen by NetMHCII 2.2 server based on artificial neuron networks (50).
Designing the multi-epitope vaccine:
The multi-epitope vaccine was designed using B cell linear epitopes, high affinity HTLs epitopes, and high scoring CTLs epitopes. These epitopes were fused together via AAY and GPGPG linkers. Moreover, a 50 S ribosomal protein L7/L12 of Mycobacterium tuberculosis (UniProt Accession no. P9WHE3) was selected as a fused adjuvant in order to boost the immunogenicity of the vaccine.
IFN-γ inducing epitopes prediction:
Interferon gamma (IFN-γ) is a central cytokine in adaptive and innate immunity which provokes macrophages and natural killer cell responses, and elevates the immune response against MHC binding epitopes. IFNepitope server (http://crdd.osdd.net/raghava/ifnepitope/scan.php) was used for prediction of the 15-mer IFN-γ epitopes of the vaccine.
Evaluation of antigenicity and allergenicity:
The chimeric vaccine antigenicity was evaluated by ANTIGENpro and VaxiJen v2.0. ANTIGENpro (http://scratch.proteomics.ics.uci.edu/) predicts antigenicity of proteins according to microarray data and provides an antigenicity index. This server has an accuracy about 76% when using a combined dataset based on cross-validation examinations (51). Further analysis of antigenicity was done by VaxiJen 2.0 server (http://www.ddgpharmfac.net/vaxijen/VaxiJen/VaxiJen.html). This server evaluates antigenicity of proteins based on the auto- and cross-covariance (ACC) transformation of protein sequences into uniform vectors of main amino acid features. These severs predict the antigenicity on the basis of various physicochemical properties of the proteins. Besides, their prediction is alignment free (52).
AllerTOP v2.0 and AllergenFP were employed to evaluate the allergenicity of the multi-epitope vaccine. AllerTOP v2.0 (http://www.ddg-pharmfac.net/AllerTOP) classifies allergens based on E-descriptors of amino acids, auto- and cross-covariance transformation, and the k nearest neighbors (kNN) machine learning methods. The accuracy of this method is 85.3% (53). Moreover, AllergenFP (http://ddg-pharmfac.net/AllergenFP/) utilizes a descriptor dependent and alignment free method to discriminate allergens and non-allergens. The method consists of a four-step algorithm. The accuracy of this method is 88% (54). The allergenicity of the chimeric vaccine was re-checked by AllergenFP.
Prediction of physiochemical properties and solubility:
ProtParam web server (http://web.expasy.org/protparam/) was utilized to evaluate the different physiochemical parameters of the multi-epitope vaccine (55). The molecular weight, theoretical isoelectric point (pI), net charge, instability index, in vitro and in vivo half-life, aliphatic index, and grand average of hydropathicity (GRAVY) of the chimeric vaccine were assessed by ProtParam. The solubility of the vaccine was predicted by SOLpro (http://scratch.proteomics.ics.uci.edu/) which is a sequence-based predictor and has an overall accuracy of 74% (56).
Homology modelling and refinement of 3D structure:
I-TASSER (Iterative Threading Assembly Refinement) server (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) was employed for homology modelling of the multi-epitope vaccine. This server predicts the structure of protein via the sequence-to-structure-to-function paradigm. Based on amino acids sequence, I-TASSER produces 3D models from multiple threading alignments and iterative structural assembly simulations (57). According to the last five community-wide CASP examinations, I-TASSER is known as the best predictor of protein structure (58). The 3D structure predicted by I-TASSER was submitted to the GalaxyRefine server (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) for model refinement. This server primarily rebuilds and repacks side chains for achievement of overall structure relaxation. Based on the CASP10 examinations, the method of GalaxyRefine server is the best for improving quality of local structure (59).
Validation of 3D structure:
It is necessary to validate the predicted models to detect probable errors of 3D models (58). ProSA-web (https://prosa.services.came.sbg.ac.at/prosa.php) was utilized primarily to compare 3D models before and after refinement. ProSA-web is an easy-to-use online program and is usually employed for validation of protein 3D structure. ProSA-web evaluates total quality of input 3D structure, and indicates quality score in comparison with all known structures for proteins. If the quality score gets out of the range of native proteins, the predicted structure probably has errors (60). Ramachandran plots for 3D structure before and after refinement were obtained through the RAMPAGE server (http://mordred.bioc.cam.ac.uk/~rapper/rampage.php) and the PROCHECK server (https://servicesn.mbi.ucla.edu/PROCHECK/). The Ramachandran plot defines energetically allowed and disallowed dihedral angles psi (ψ) and phi (ϕ) of protein amino acids, and is calculated according to van der Waal radius of the residues side chains. The results of RAMPAGE and PROCHECK indicate the percentage of residues in allowed and disallowed regions which shows predicted model quality (61).
Prediction of discontinuous B-cell epitope:
It is indicated that more than 90% of B cell epitopes of antigens are discontinuous (conformational). These epitopes are separated parts of an antigen sequence which are placed next to each other when protein folds (62). Discontinuous B cell epitopes of the multi-epitope vaccine were predicted by ElliPro (http://tools.iedb.org/ellipro/). ElliPro utilizes three algorithms to draw the structure of a protein as an ellipsoid, analyze the residue protrusion index (PI), and cluster close residues based on their PI values. Each output epitope of ElliPro has a score which is defined as an average PI value for every residue of epitope. For example, a PI value of 0.9 shows 90% of protein residues reside in the ellipsoid, while the remaining 10% are out of the ellipsoid (63).
Molecular docking of the vaccine and TLR4 :
In this work, HADDOCK 2.4 web server (https://bianca.science.uu.nl/haddock2.4) was used for molecular docking of the multi-epitope vaccine against the TLR4 (PDB ID: 4g8a) receptor. HADDOCK is a server for prediction of protein-protein interaction based on biochemical or biophysical information. At first, preparation of their PBD is done, so job is submitted on HADDOCK 2.4 and ran. After analysis, 11 clusters were obtained and the best structure was selected according to RMSD and binding energy.
Elastic-mode analysis:
Elastic-mode study is very important to evaluate the stability of the protein-protein complex in computational analysis. The iMODS server (http://imods.chaconlab.org/) was utilized to describe the sum of protein motion in the internal coordinates through normal mode analysis (NMA). The server predicted the direction and extent of the immanent motions of the complex in terms of deformability, eigenvalues, B-factors, and covariance. The deformability of the core chain depends on whether a specified molecule can deform at every residue. The eigenvalue of each normal mode indicates the rigidity of motion. This value is related directly to the energy required for the structural deformation, and the deformation is much easier if the eigenvalue is low (64).
Molecular dynamic simulation (MDS):
The molecular dynamic simulation (MDS) is used to check the stability of protein-ligand complex. It was conducted with NAMD2 software after docking TLR4 and SARS-Cov2 spike protein to overview the system dynamics in an aqueous solution. NAMDs are numerically able to estimate atomic trajectories by solving equations of motion. CHARMM force field as an empirical force field approximates the actual atomic force in biopolymer systems to compute atomic trajectories. This simulation protocol was started with 20,000 minimization steps, and each step was one fs, and the total steps were 4,500,000 and temperature set up was on 300 K. After simulation, the achieved trajectory file analysis was carried out to check flexibility, compactness, stability of purposed vaccine structure during 4.5 nanoseconds simulation time with VMD.
mRNA secondary structure prediction:
Mfold online software was employed to predict the mRNA secondary structure of the favored chimeric vaccine containing minimum ΔG positive base pairs using thermodynamic methods. The circle graph was also used to better show the interaction between the base pairs. Indeed, it also facilitates to find a structure with the minimum overlap of bases. The graph places the bases equally around a circle in a clockwise fashion and draws an arc between paired bases. Intersecting arcs represent Pseudoknots.
DNA optimization and in silico cloning:
Every living organism uses different types of codons for same amino acids which is called codon bias. When a recombinant protein is going to be produced by a foreign host, the codons of its DNA should be optimized according to the host. Codon optimization might increase expression levels up to more than 1000 folds (65). Using JCAT online server (http://www.jcat.de) the amino acid sequence of the designed vaccine was reverse translated into the nucleotide sequence. Furthermore, GenScript Rare Codon Analysis Tool (https://www.genscript.com/tools/rare-codon-analysis) was used to optimize the codons for efficient overexpression in Escherichia coli (E. coli). To construct the recombinant vector, the optimized sequence was cloned into pET-28a (+) expression vector between NcoI and XhoI sites using SnapGene 5.1.2 software.
Results
Linear B cell epitopes:
Linear B-cell epitopes with different length were predicted by ABCpred and BepiPred-2.0. Only those epitopes that were predicted by both servers or had a considerable overlapping were picked, and then ten Linear B-cell epitopes were chosen for further analysis (Table 1).
Coronaviruses (CoVs) are a member of the subfamily Othocoronavirinae, in the family Coronaviridae (1-3). They are extremely pathogenic and are often known to be infectious, as shown by the 2002 and 2012 epidemic of severe acute respiratory syndrome (SARS) and middle east respiratory syndrome (MERS) (4, 5). In late 2019, an outbreak of another coronavirus that triggers a respiratory-related disease was identified in Wuhan, Hubei, China, a disease now publicly named “the Corona Virus Disease 2019; COVID-19” (6). Hence, World Health Organization (WHO) announced global health disaster and regarded the COVID-19 as pandemic (7, 8). The major clinical symptoms in patients with SARS-CoV-2 are fatigue, cough, trouble breathing, shortness of breath, increased levels of lactate dehydrogenase (LDH), and extended prothrombin period. The less common signs involved sputum development, headache, hemoptysis, and diarrhea (9, 10). Genome-wide phylogenetic study reveals that SARS-CoV-2 shares 79.5%, 50%, and 96% sequence similarity with SARS-CoV, MERS-CoV, and bat coronavirus, respectively (11). Unlike other coronaviruses, the new SARS-CoV-2 is a positive-sense single-stranded RNA virus. Moreover, its genome size ranges between about 26,000 and 32,000 bases, comprises a variable number of open reading frames (ORFs). This novel virus is spherical with a diameter of approximately 125 nm, as well as it encodes 9860 amino acids translating into many non-structural proteins such as replicas (orf1a / b), nsp2, nsp3 and accessory proteins (orf3a, orf7a / b) along with structural proteins including spike (S), envelope (E), membrane (M), and nucleocapsid (N) proteins (12-14). Of these structural proteins, the spike glycoprotein was shown to attach to the cellular receptor angiotensin-converting enzyme 2 (ACE2), facilitating the virus entry to the host cells (15-18). Structurally, the S protein belongs to the class I viral protein, which contains two functional subunits, an amino-terminal S1 (685 aa) and a carboxyl-terminal S2 (588 aa) subunit (19). Each of these two subunits has a specific task. The subunit S1 plays a critical role in the virus binding to the host cell receptors, while the subunit S2 is involved in virus-host membrane fusion. It was found that the S1 protein of SARS-CoV-2 shows approximately 70% similarity with that of human SARS-CoVs. This subunit has two main domains: I) an N-terminal domain (NTD) that mediates sugar binding, II) a C-terminal domain (CTD) that facilitates protein receptor recognition. Therefore, both domains are considered as receptor-binding domains (RBDs) (20-22). Numerous data indicate that the S protein, as a virus key antigen, plays a crucial role in stimulating the production of neutralizing antibodies which accordingly inhibit the virus entry to the host cells. Hence, blocking the initial entry of a virus is suggested as a valuable approach in managing various viral diseases (21, 23, 24).
Currently, there are several nonspecific approaches for the control of SARS-CoV-2, including administration of type I IFNs, ribavirin, lopinavir/ritonavir, remdesivir, nelfinavir, arbidol, Chloroquine, and convalescent plasma (25, 26). Although vaccination is regarded the most efficient modality in prevention of infection diseases, yet there is no approved specific vaccine for SARS-CoV-2 (25). (27-29).
Today, bioinformatics and computational biology have influenced various aspects of biology and medicine (30-32). Using computational approaches prior to beginning an experimental study, minimizes the costs and time while improving the precision and validity of the experimental studies (33-35). In recent years, in silico approaches have enabled us to design epitope-based vaccine against various diseases such as infectious diseases and cancers. These approaches speed up the process of designing, developing, and evaluation of vaccine candidates (32, 36-38). Multi-epitope vaccines are constructed utilizing the most immunodominant regions (components) of the pathogen's proteins which have conserved sequences that allows them more success and can support in opposing the high mutation frequency in RNA viruses. These types of vaccines have numerous advantages such as safety, efficiency, simple production in large-scale, cost-curtailing, antigenicity, and immunogenicity (39-42). An optimal vaccine should include both B-cell and T-cell epitopes, for which the vaccine may effectively either induce particular humoral and cellular immunity against pathogens (43). The production of antibodies by B cells activation is essential for maintaining immunity to coronavirus, along with the virus-specific killer and memory CD8 + T cells to eradicate infected cells. Given the contribution of multi-epitope vaccines in responses restricted via a broad range of HLA molecules and make a balanced CD4+ and CD8+ cellular immune response, this strategy can be useful in clinic (44, 45).
We decided to design a multi-epitope vaccine against SARS-CoV-2 based on the virus S protein, and evaluated its biochemical and immune stimulatory features using in silico approaches.
.
Materials & Methods
Antigen selection:
Recently, the genome of SARS-CoV-2 was sequenced and the proteins coded by the virus were identified. The amino acid sequence of spike (S) protein was picked up from NCBI Protein database under reference number: YP_009724390.1. This protein is 1273 amino acids long.
Prediction of linear B cell epitopes:
The B-cell epitopes of antigens are recognized by humoral immune system. This type of epitope is the specific part of the antigen which is a target for B cell receptors. These epitopes have an essential role in designing an efficient vaccine. Linear B-cell epitopes were evaluated mostly by the BepiPred-2.0 web server (http://www.cbs.dtu.dk/services/BepiPred/) and ABCpred prediction server (http://crdd.osdd.net/raghava/abcpred/). Since BepiPred-2.0 is a strict server, the threshold was considered 0.51. BepiPred-2.0 predicts according to a random forest algorithm which is expert for identifying epitopes from interactions of antibody-antigen. 14-mers epitopes were anticipated by ABCpred with a threshold of 0.85 because this server is more lenient. ABCpred predicts epitopes based on recurrent neural network method (46, 47).
Prediction of cytotoxic T lymphocytes (CTL) epitopes:
Cytotoxic T lymphocyte (CTL) epitopes of S antigen were evaluated via NetCTL 1.2 server (http://www.cbs.dtu.dk/services/NetCTL/). This server method contains combination of MHC I binding peptides, cleavage of C-terminus in proteasomes, and TAP (Transporter Associated with Antigen Processing) transport efficacy predictions. However, the prediction of CTL epitopes in this server is limited to 12-mers peptides for MHC I supertypes. In this study, only the HLA-A2 was chosen. The prediction of MHC I binding and peptide cleavage in proteasomes was carried out by artificial neural networks. TAP transportation was evaluated by a weight matrix (48). The predictions were sorted based on combined score. The threshold for the prediction of CTL epitopes was set at 0.75 (default). The sequence of the S antigen was submitted to CTLPred server (http://crdd.osdd.net/raghava/ctlpred/). The prediction method of CTLPred is depended on a sophisticated machine learning techniques called artificial neural network (ANN) and support vector machine (SVM) (49). The combined prediction method was selected for this study. The thresholds were at default scores.
Helper T-lymphocyte (HTL) epitopes prediction:
Prediction of 15-mers HTL epitopes for human alleles was carried out using NetMHCII 2.3 Server (http://www.cbs.dtu.dk/services/NetMHCII/). Epitope peptides that bind HLA-DR, HLA-DQ, and HLA-DP alleles of human were chosen by NetMHCII 2.2 server based on artificial neuron networks (50).
Designing the multi-epitope vaccine:
The multi-epitope vaccine was designed using B cell linear epitopes, high affinity HTLs epitopes, and high scoring CTLs epitopes. These epitopes were fused together via AAY and GPGPG linkers. Moreover, a 50 S ribosomal protein L7/L12 of Mycobacterium tuberculosis (UniProt Accession no. P9WHE3) was selected as a fused adjuvant in order to boost the immunogenicity of the vaccine.
IFN-γ inducing epitopes prediction:
Interferon gamma (IFN-γ) is a central cytokine in adaptive and innate immunity which provokes macrophages and natural killer cell responses, and elevates the immune response against MHC binding epitopes. IFNepitope server (http://crdd.osdd.net/raghava/ifnepitope/scan.php) was used for prediction of the 15-mer IFN-γ epitopes of the vaccine.
Evaluation of antigenicity and allergenicity:
The chimeric vaccine antigenicity was evaluated by ANTIGENpro and VaxiJen v2.0. ANTIGENpro (http://scratch.proteomics.ics.uci.edu/) predicts antigenicity of proteins according to microarray data and provides an antigenicity index. This server has an accuracy about 76% when using a combined dataset based on cross-validation examinations (51). Further analysis of antigenicity was done by VaxiJen 2.0 server (http://www.ddgpharmfac.net/vaxijen/VaxiJen/VaxiJen.html). This server evaluates antigenicity of proteins based on the auto- and cross-covariance (ACC) transformation of protein sequences into uniform vectors of main amino acid features. These severs predict the antigenicity on the basis of various physicochemical properties of the proteins. Besides, their prediction is alignment free (52).
AllerTOP v2.0 and AllergenFP were employed to evaluate the allergenicity of the multi-epitope vaccine. AllerTOP v2.0 (http://www.ddg-pharmfac.net/AllerTOP) classifies allergens based on E-descriptors of amino acids, auto- and cross-covariance transformation, and the k nearest neighbors (kNN) machine learning methods. The accuracy of this method is 85.3% (53). Moreover, AllergenFP (http://ddg-pharmfac.net/AllergenFP/) utilizes a descriptor dependent and alignment free method to discriminate allergens and non-allergens. The method consists of a four-step algorithm. The accuracy of this method is 88% (54). The allergenicity of the chimeric vaccine was re-checked by AllergenFP.
Prediction of physiochemical properties and solubility:
ProtParam web server (http://web.expasy.org/protparam/) was utilized to evaluate the different physiochemical parameters of the multi-epitope vaccine (55). The molecular weight, theoretical isoelectric point (pI), net charge, instability index, in vitro and in vivo half-life, aliphatic index, and grand average of hydropathicity (GRAVY) of the chimeric vaccine were assessed by ProtParam. The solubility of the vaccine was predicted by SOLpro (http://scratch.proteomics.ics.uci.edu/) which is a sequence-based predictor and has an overall accuracy of 74% (56).
Homology modelling and refinement of 3D structure:
I-TASSER (Iterative Threading Assembly Refinement) server (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) was employed for homology modelling of the multi-epitope vaccine. This server predicts the structure of protein via the sequence-to-structure-to-function paradigm. Based on amino acids sequence, I-TASSER produces 3D models from multiple threading alignments and iterative structural assembly simulations (57). According to the last five community-wide CASP examinations, I-TASSER is known as the best predictor of protein structure (58). The 3D structure predicted by I-TASSER was submitted to the GalaxyRefine server (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) for model refinement. This server primarily rebuilds and repacks side chains for achievement of overall structure relaxation. Based on the CASP10 examinations, the method of GalaxyRefine server is the best for improving quality of local structure (59).
Validation of 3D structure:
It is necessary to validate the predicted models to detect probable errors of 3D models (58). ProSA-web (https://prosa.services.came.sbg.ac.at/prosa.php) was utilized primarily to compare 3D models before and after refinement. ProSA-web is an easy-to-use online program and is usually employed for validation of protein 3D structure. ProSA-web evaluates total quality of input 3D structure, and indicates quality score in comparison with all known structures for proteins. If the quality score gets out of the range of native proteins, the predicted structure probably has errors (60). Ramachandran plots for 3D structure before and after refinement were obtained through the RAMPAGE server (http://mordred.bioc.cam.ac.uk/~rapper/rampage.php) and the PROCHECK server (https://servicesn.mbi.ucla.edu/PROCHECK/). The Ramachandran plot defines energetically allowed and disallowed dihedral angles psi (ψ) and phi (ϕ) of protein amino acids, and is calculated according to van der Waal radius of the residues side chains. The results of RAMPAGE and PROCHECK indicate the percentage of residues in allowed and disallowed regions which shows predicted model quality (61).
Prediction of discontinuous B-cell epitope:
It is indicated that more than 90% of B cell epitopes of antigens are discontinuous (conformational). These epitopes are separated parts of an antigen sequence which are placed next to each other when protein folds (62). Discontinuous B cell epitopes of the multi-epitope vaccine were predicted by ElliPro (http://tools.iedb.org/ellipro/). ElliPro utilizes three algorithms to draw the structure of a protein as an ellipsoid, analyze the residue protrusion index (PI), and cluster close residues based on their PI values. Each output epitope of ElliPro has a score which is defined as an average PI value for every residue of epitope. For example, a PI value of 0.9 shows 90% of protein residues reside in the ellipsoid, while the remaining 10% are out of the ellipsoid (63).
Molecular docking of the vaccine and TLR4 :
In this work, HADDOCK 2.4 web server (https://bianca.science.uu.nl/haddock2.4) was used for molecular docking of the multi-epitope vaccine against the TLR4 (PDB ID: 4g8a) receptor. HADDOCK is a server for prediction of protein-protein interaction based on biochemical or biophysical information. At first, preparation of their PBD is done, so job is submitted on HADDOCK 2.4 and ran. After analysis, 11 clusters were obtained and the best structure was selected according to RMSD and binding energy.
Elastic-mode analysis:
Elastic-mode study is very important to evaluate the stability of the protein-protein complex in computational analysis. The iMODS server (http://imods.chaconlab.org/) was utilized to describe the sum of protein motion in the internal coordinates through normal mode analysis (NMA). The server predicted the direction and extent of the immanent motions of the complex in terms of deformability, eigenvalues, B-factors, and covariance. The deformability of the core chain depends on whether a specified molecule can deform at every residue. The eigenvalue of each normal mode indicates the rigidity of motion. This value is related directly to the energy required for the structural deformation, and the deformation is much easier if the eigenvalue is low (64).
Molecular dynamic simulation (MDS):
The molecular dynamic simulation (MDS) is used to check the stability of protein-ligand complex. It was conducted with NAMD2 software after docking TLR4 and SARS-Cov2 spike protein to overview the system dynamics in an aqueous solution. NAMDs are numerically able to estimate atomic trajectories by solving equations of motion. CHARMM force field as an empirical force field approximates the actual atomic force in biopolymer systems to compute atomic trajectories. This simulation protocol was started with 20,000 minimization steps, and each step was one fs, and the total steps were 4,500,000 and temperature set up was on 300 K. After simulation, the achieved trajectory file analysis was carried out to check flexibility, compactness, stability of purposed vaccine structure during 4.5 nanoseconds simulation time with VMD.
mRNA secondary structure prediction:
Mfold online software was employed to predict the mRNA secondary structure of the favored chimeric vaccine containing minimum ΔG positive base pairs using thermodynamic methods. The circle graph was also used to better show the interaction between the base pairs. Indeed, it also facilitates to find a structure with the minimum overlap of bases. The graph places the bases equally around a circle in a clockwise fashion and draws an arc between paired bases. Intersecting arcs represent Pseudoknots.
DNA optimization and in silico cloning:
Every living organism uses different types of codons for same amino acids which is called codon bias. When a recombinant protein is going to be produced by a foreign host, the codons of its DNA should be optimized according to the host. Codon optimization might increase expression levels up to more than 1000 folds (65). Using JCAT online server (http://www.jcat.de) the amino acid sequence of the designed vaccine was reverse translated into the nucleotide sequence. Furthermore, GenScript Rare Codon Analysis Tool (https://www.genscript.com/tools/rare-codon-analysis) was used to optimize the codons for efficient overexpression in Escherichia coli (E. coli). To construct the recombinant vector, the optimized sequence was cloned into pET-28a (+) expression vector between NcoI and XhoI sites using SnapGene 5.1.2 software.
Results
Linear B cell epitopes:
Linear B-cell epitopes with different length were predicted by ABCpred and BepiPred-2.0. Only those epitopes that were predicted by both servers or had a considerable overlapping were picked, and then ten Linear B-cell epitopes were chosen for further analysis (Table 1).
Table 1. Consensus linear B cell epitopes predicted by ABCpred and BepiPred-2.0
| Peptide | Consensus epitope | Start position | End position |
| BE1 | QCVNLTTRTQLPPAYTNSFTRGVYY | 14 | 38 |
| BE2 | IAPGQTGKIADYNYKLPDDF | 410 | 429 |
| BE3 | SASFSTFKCYGVSPTKLNDLCFT | 371 | 393 |
| BE4 | AVEQDKNTQEVFAQ | 771 | 784 |
| BE5 | FRKSNLKPFERDISTEIYQAGSTPCNGVEGFNCYFPL | 455 | 492 |
| BE6 | FSNVTWFHAIHVSGTNGTKRFDNPV | 59 | 83 |
| BE7 | SFELLHAPATVCGPKKSTNLV | 514 | 534 |
| BE8 | PNITNLCPFGEVFNATRFASVYAWNRKRI | 330 | 358 |
| BE9 | DQLTPTWRVYSTGSNVFQTRAGCLIGAEHVNNSYECDI | 627 | 664 |
| BE10 | SYQTQTNSPRRARSVASQSIIAYTMSLGAENSVAYSN | 673 | 710 |
Cytotoxic T lymphocytes (CTL) epitopes:
NetCTL 1.2 and CTLPred servers predicted 42 and 3 CTL (9-mer) epitopes within the structure, respectively. Eight CTL epitopes possessing the highest scores were finally selected based on the results of both servers (Table 2).
NetCTL 1.2 and CTLPred servers predicted 42 and 3 CTL (9-mer) epitopes within the structure, respectively. Eight CTL epitopes possessing the highest scores were finally selected based on the results of both servers (Table 2).
Table 2. High scored and consensus cytotoxic T cells (CTL) epitopes predicted by NetCTL and CTLPred
| Peptide | CTL epitopes | Start position | End position | NetCTL score | CTLPred (ANN/SVM) score |
| CE1 | YLQPRTFLL | 269 | 278 | 1.5152 | |
| CE2 | KIADYNYKL | 417 | 426 | 1.4347 | |
| CE3 | SIIAYTMSL | 691 | 700 | 1.3658 | |
| CE4 | VLNDILSRL | 976 | 985 | 1.3533 | |
| CE5 | RLDKVEAEV | 983 | 992 | 1.0612 | 0.71/1.6892505 |
| CE6 | RLQSLQTYV | 1000 | 1009 | 1.2727 | |
| CE7 | NLNESLIDL | 1192 | 1201 | 0.9825 | 0.92/1.5879094 |
| CE8 | FIAGLIAIV | 1220 | 1229 | 1.2124 |
Helper T Lymphocytes (HTL) epitopes:
NetMHCII 2.2 server predicted MHC-II binding epitopes with high reliability (also called HTL epitopes) which had the most tendency for human alleles HLA-DR, HLA-DQ, and HLA-DP. Three epitopes with highest scores were chosen for each allele. Finally, nine (15-mer) epitopes were selected as HTL epitopes (Table 3).
NetMHCII 2.2 server predicted MHC-II binding epitopes with high reliability (also called HTL epitopes) which had the most tendency for human alleles HLA-DR, HLA-DQ, and HLA-DP. Three epitopes with highest scores were chosen for each allele. Finally, nine (15-mer) epitopes were selected as HTL epitopes (Table 3).
Table 3. Highest scored helper T cells (HTL) epitopes predicted by NetMHCII 2.2.
| Peptide | Peptide epitope | Allele | Reliability |
| HE1 | PVAIHADQLTPTWRV | HLA-DR | 0.96 |
| HE2 | RAAEIRASANLAATK | HLA-DR | 0.94 |
| HE3 | RGVYYPDKVFRSSVL | HLA-DR | 0.94 |
| HE4 | FGEVFNATRFASVYA | HLA-DP | 0.88 |
| HE5 | LIRAAEIRASANLAA | HLA-DP | 0.62 |
| HE6 | YFKIYSKHTPINLVR | HLA-DP | 0.58 |
| HE7 | TQQLIRAAEIRASAN | HLA-DQ | 0.81 |
| HE8 | TGSNVFQTRAGCLIG | HLA-DQ | 0.72 |
| HE9 | QLTPTWRVYSTGSNV | HLA-DQ | 0.67 |
Multi-epitope vaccine design:
Totally, 10 linear B cell, nine HTL, and eight CTL epitopes were used to construct the multi-epitope vaccine. Linear B cell and HTL epitopes were fused by GPGPG linkers, while CTL epitopes were linked together via AAY linkers. 50S ribosomal L7/L12 protein, a TLR4 agonist, was used as an adjuvant which was added to the N-terminus of the vaccine by an EAAAK linker. A hexa histidine tag (His tag) was also added to the C-terminus for purification purpose. The final designed recombinant multi-epitope vaccine consisted of 731 amino acids (Figure 1).
Totally, 10 linear B cell, nine HTL, and eight CTL epitopes were used to construct the multi-epitope vaccine. Linear B cell and HTL epitopes were fused by GPGPG linkers, while CTL epitopes were linked together via AAY linkers. 50S ribosomal L7/L12 protein, a TLR4 agonist, was used as an adjuvant which was added to the N-terminus of the vaccine by an EAAAK linker. A hexa histidine tag (His tag) was also added to the C-terminus for purification purpose. The final designed recombinant multi-epitope vaccine consisted of 731 amino acids (Figure 1).
Fig 1. The multi-epitope vaccine construct. The adjuvant resides on N-terminus (green). BEs, HEs, and CE represent B cell linear epitopes, helper T cell epitopes, and cytotoxic T cell epitopes, respectively. The His-tag for affinity purification is located on C-terminus.
IFN-γ induction:
Based on the results of IFNepitope server, 122 potential IFN-γ inducing peptides (with both negative and positive scores) were predicted for being as the adjuvant. Sixty-two peptides possessed positive scores. Moreover, within the main part of the designed vaccine, 279 probable IFN-γ inducing peptides were identified among which 87 were positive. Totally, 149 positive IFN-γ inducing peptides were predicted in the structure of the multi-epitope vaccine.
Antigenicity and allergenicity:
According to the results of VaxiJen 2.0 and ANTIGENpro servers, the antigenicity score was 0.4606 (threshold: 0.4) and 0.835593 for the vaccine (whole construct including the adjuvant), respectively. The results showed that the designed multi-epitope vaccine could be an antigen for human body and elicit immune responses. The results of AllerTOP v.2.0 and AllergenFP v.1.0 servers indicated that the multi-epitope could be a probable non-allergen protein in human body.
Physiochemical properties and solubility:
Based on the results of ProtParam, the molecular weight (MW), theoretical pI, and total charge for the designed vaccine were 76863.88 Da, 8.66, and +7, respectively. The positive charge and the pH of the chimeric vaccine indicate that it is a basic protein. The half-life of the protein was evaluated to be 30 hours in mammalian reticulocyte cells in vitro, more than 20 hours in yeast in vivo, and more than 10 hours in E. coli in vivo. The instability index for the vaccine was 20.46 indicating that the vaccine could be considered as a stable protein. The predicted aliphatic index was 76.18, which means that the vaccine could be a thermostable protein. The estimated grand average of hydropathicity (GRAVY) was -0.164. The negative value means that the protein is hydrophilic and could interact with water molecules (58). SOLpro estimated that the designed vaccine would be a soluble protein upon overexpression with a probability of 0.920822.
3D structure modelling and refinement:
Five 3D structure models were predicted for the designed multi-epitope vaccine by I-TASSER server based on 10 threading templates. The predicted models had C-scores ranging from -3.68 to -1.15. The C-score usually has a range between −5 and 2, in which higher values show more confidence. Model 1 was chosen for further refinement process due to highest C-score (Figure 2A). The chosen model had an assessed TM-score of 0.57 ± 0.15 with an estimated RMSD of 10.9 ± 4.6 Å. The TM-score is used as a scale for determining the structural similarity between two structures. The TM-score was suggested to overwhelm the problem of RMSD, which is sensitive to local error. A TM-score more than 0.5 shows correct topology in the model, while a TM-score less than 0.17 indicates random similarity. These cut-offs of TM-score are independent of protein length (58). The primary chosen model was refined by the GalaxyRefine server which generated five refined model. For model 5 the Rama favored residues before and after refinement were 62.6 and 85.0, respectively. This model was chosen as final model for further analysis. The selected unrefined and refined model is shown in figure 2 A and B.
Based on the results of IFNepitope server, 122 potential IFN-γ inducing peptides (with both negative and positive scores) were predicted for being as the adjuvant. Sixty-two peptides possessed positive scores. Moreover, within the main part of the designed vaccine, 279 probable IFN-γ inducing peptides were identified among which 87 were positive. Totally, 149 positive IFN-γ inducing peptides were predicted in the structure of the multi-epitope vaccine.
Antigenicity and allergenicity:
According to the results of VaxiJen 2.0 and ANTIGENpro servers, the antigenicity score was 0.4606 (threshold: 0.4) and 0.835593 for the vaccine (whole construct including the adjuvant), respectively. The results showed that the designed multi-epitope vaccine could be an antigen for human body and elicit immune responses. The results of AllerTOP v.2.0 and AllergenFP v.1.0 servers indicated that the multi-epitope could be a probable non-allergen protein in human body.
Physiochemical properties and solubility:
Based on the results of ProtParam, the molecular weight (MW), theoretical pI, and total charge for the designed vaccine were 76863.88 Da, 8.66, and +7, respectively. The positive charge and the pH of the chimeric vaccine indicate that it is a basic protein. The half-life of the protein was evaluated to be 30 hours in mammalian reticulocyte cells in vitro, more than 20 hours in yeast in vivo, and more than 10 hours in E. coli in vivo. The instability index for the vaccine was 20.46 indicating that the vaccine could be considered as a stable protein. The predicted aliphatic index was 76.18, which means that the vaccine could be a thermostable protein. The estimated grand average of hydropathicity (GRAVY) was -0.164. The negative value means that the protein is hydrophilic and could interact with water molecules (58). SOLpro estimated that the designed vaccine would be a soluble protein upon overexpression with a probability of 0.920822.
3D structure modelling and refinement:
Five 3D structure models were predicted for the designed multi-epitope vaccine by I-TASSER server based on 10 threading templates. The predicted models had C-scores ranging from -3.68 to -1.15. The C-score usually has a range between −5 and 2, in which higher values show more confidence. Model 1 was chosen for further refinement process due to highest C-score (Figure 2A). The chosen model had an assessed TM-score of 0.57 ± 0.15 with an estimated RMSD of 10.9 ± 4.6 Å. The TM-score is used as a scale for determining the structural similarity between two structures. The TM-score was suggested to overwhelm the problem of RMSD, which is sensitive to local error. A TM-score more than 0.5 shows correct topology in the model, while a TM-score less than 0.17 indicates random similarity. These cut-offs of TM-score are independent of protein length (58). The primary chosen model was refined by the GalaxyRefine server which generated five refined model. For model 5 the Rama favored residues before and after refinement were 62.6 and 85.0, respectively. This model was chosen as final model for further analysis. The selected unrefined and refined model is shown in figure 2 A and B.
Fig 2. (A) 3D model before refinement. (B) 3D model after refinement. (C) Z-score before 3D model refinement. (D) Z-score after 3D model refinement.
3D structure validation
The Ramachandran plot analysis of model before and after refinement was comprised by RAMPAGE and PROCHECK servers. The percent of residues in favored region were considerably increased (Table 4). The ProSA-web results showed a Z-Score of -3.15 for the primary model and Z-Score of -3.55 for the refined model (Figure 2 C and D). The model resided outside the score range is usually seen for native proteins of comparable size.
The Ramachandran plot analysis of model before and after refinement was comprised by RAMPAGE and PROCHECK servers. The percent of residues in favored region were considerably increased (Table 4). The ProSA-web results showed a Z-Score of -3.15 for the primary model and Z-Score of -3.55 for the refined model (Figure 2 C and D). The model resided outside the score range is usually seen for native proteins of comparable size.
Table 4. Comparison of residues residence in Ramachandran plot before and after model refinement.
| Before refinement | After refinement | ||
RAMPAGE |
Number of residues in favored region | 481 (66.0%) | 632 (86.9%) |
| Number of residues in allowed region | 152 (20.9%) | 72 (9.9%) | |
| Number of residues in outlier region | 96 (13.2%) | 25 (3.4%) | |
PROCHECK |
Residues in most favored regions | 301 (51.5%) | 435 (74.5%) |
| Residues in additional allowed regions | 230 (39.4%) | 120 (20.5%) | |
| Residues in generously allowed regions | 37 (6.3%) | 15 (2.6%) | |
| Residues in disallowed regions | 16 (2.7%) | 14 (2.4%) |
Discontinuous B-cell epitopes
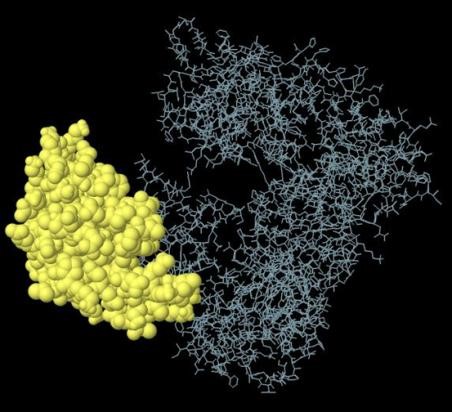
A total of seven discontinuous B cell epitopes were estimated to be present in the designed multi-epitope vaccine with scores ranging from 0.527 to 0.754. The conformational epitopes ranged in size from 4 to 115 residues (Table 5). The largest discontinuous B cell epitope is shown in figure 3.
A total of seven discontinuous B cell epitopes were estimated to be present in the designed multi-epitope vaccine with scores ranging from 0.527 to 0.754. The conformational epitopes ranged in size from 4 to 115 residues (Table 5). The largest discontinuous B cell epitope is shown in figure 3.
Fig 3. The biggest discontinuous B cell epitope of the multi-epitope vaccine.
Table 5. Discontinuous B cell epitopes of multi-epitope vaccine.
Table 5. Discontinuous B cell epitopes of multi-epitope vaccine.
| Number | Residues | Number of residues | Score |
| 1 | A:M1, A:A2, A:K3, A:L4, A:S5, A:T6, A:D7, A:E8, A:L9, A:L10, A:D11, A:A12, A:F13, A:K14, A:E15, A:M16, A:T17, A:L18, A:L19, A:E20, A:L21, A:S22, A:D23, A:F24, A:V25, A:K26, A:K27, A:F28, A:E29, A:E30, A:T31, A:F32, A:E33, A:V34, A:T35, A:A36, A:A37, A:A38, A:P39, A:V40, A:A41, A:V42, A:A43, A:A44, A:A45, A:G46, A:A47, A:A48, A:P49, A:A50, A:G51, A:A52, A:A53, A:V54, A:E55, A:A56, A:A57, A:E58, A:E59, A:Q60, A:S61, A:E62, A:F63, A:D64, A:V65, A:I66, A:L67, A:E68, A:A69, A:A70, A:G71, A:D72, A:K73, A:K74, A:I75, A:G76, A:V77, A:I78, A:K79, A:V80, A:V81, A:S86, A:G87, A:L88, A:L90, A:K91, A:E92, A:A93, A:K94, A:D95, A:L96, A:V97, A:D98, A:G99, A:A100, A:P101, A:K102, A:P103, A:L104, A:L105, A:E106, A:K107, A:V108, A:A109, A:K110, A:K119, A:L120, A:E121, A:A122, A:A123, A:G124, A:A125, A:T126, A:V127, A:T128 | 115 | 0.754 |
| 2 | A:S195, A:T196, A:F197, A:K198, A:C199, A:A219, A:V220, A:E221, A:Q222, A:D223, A:K224, A:N225, A:T226, A:Q227, A:E228, A:V229, A:F230, A:A231, A:Q232, A:G233, A:P234, A:G235, A:P236, A:G237, A:F238, A:R239, A:S241, A:N242, A:L243, A:K244, A:P245, A:F246, A:E247, A:R248, A:D249, A:I250, A:S251, A:T252, A:E253, A:I254, A:Y255, A:Q256, A:A257, A:G258, A:S259, A:T260, A:P261, A:C262, A:N263, A:G264, A:V265, A:E266, A:G267, A:F268, A:N269, A:T284, A:W285, A:F286, A:H287, A:A288, A:I289, A:H290, A:V291, A:S292, A:G293, A:T294, A:N295, A:G296, A:T297, A:S310, A:E312, A:L313, A:L314, A:H315, A:A316, A:P317, A:A318, A:T319, A:V320, A:C321, A:G322, A:P323, A:K324, A:K325, A:S326, A:C342, A:P343, A:F344, A:G345, A:E346, A:V347, A:F348, A:N349, A:A350, A:T351, A:R352, A:F353 | 97 | 0.708 |
| 3 | A:R585, A:A586, A:S587, A:A588, A:N589, A:G590, A:P591, A:G592, A:P593, A:G594, A:T595, A:G596, A:S597, A:N598, A:V599, A:F600, A:Q601, A:T602, A:R603, A:A604, A:G605, A:C606, A:L607, A:I608, A:G609, A:G610, A:P611, A:G612, A:P613, A:G614, A:Q615, A:L616, A:T617, A:P618, A:T619, A:W620, A:R621, A:V622, A:Y623, A:Y700, A:V701, A:Y704, A:N705, A:L706, A:N707, A:S709, A:L710, A:I711 | 48 | 0.674 |
| 4 | A:K362, A:I364, A:W376, A:R377, A:V378, A:Y379, A:S380, A:T381, A:G382, A:R389, A:A390, A:G391, A:C392, A:L393, A:I394, A:G395, A:E397, A:G408, A:P409, A:G410, A:P411, A:G412, A:S413, A:Y414, A:Q415, A:T416, A:R422, A:R423, A:A424, A:S426, A:V427, A:A428, A:A483, A:N484, A:L485, A:A486, A:A487, A:T488, A:K489, A:G490, A:P491, A:G492, A:G494, A:R495, A:V497, A:Y498, A:Y499, A:P500, A:D501, A:K502, A:V503, A:F504, A:R505, A:S506, A:S507, A:V508, A:L509, A:G510, A:P511, A:G512, A:P513, A:G514, A:F515, A:G516, A:E517, A:V518, A:F519, A:N520, A:A521, A:T522, A:R523, A:G532, A:P533, A:G534, A:L535, A:I536, A:A538, A:A539, A:E540, A:I541, A:R542, A:A543, A:S544, A:A545, A:N546, A:L547, A:A548, A:A549, A:G550, A:P551, A:G552, A:P553, A:G554, A:Y555, A:K557, A:S560, A:K561, A:H562, A:T563, A:P564, A:I565, A:N566, A:L567, A:V568, A:R569, A:Q576, A:Q577, A:L578, A:I579, A:R580, A:A581, A:A582, A:E583, A:I584, A:S624, A:T625, A:G626, A:S627, A:N628, A:V629, A:A630, A:A631, A:Y632, A:Y633, A:F717, A:I718, A:A719, A:G720, A:L721, A:I722, A:A723, A:I724, A:V725, A:H726, A:H727, A:H728, A:H729, A:H730, A:H731 | 139 | 0.657 |
| 5 | A:I658, A:I659, A:A660, A:Y661, A:T662, A:M663, A:S664, A:L665, A:A666, A:A667 | 10 | 0.614 |
| 6 | A:L372, A:F386, A:Q387, A:T388 | 4 | 0.536 |
| 7 | A:F212, A:T213, A:G214, A:P215 | 4 | 0.527 |
Docking of the designed vaccine against TLR4 receptor:
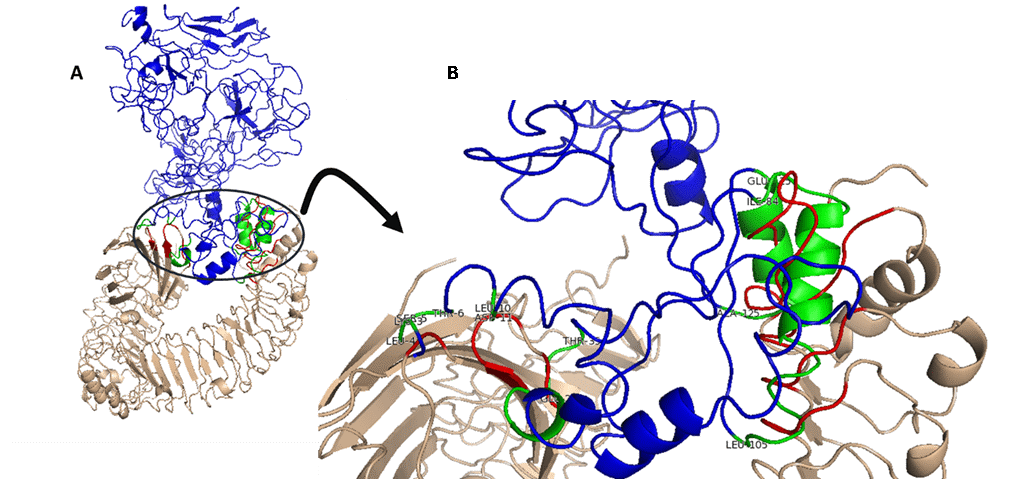
In the present study, HADDOCK server was used to predict the binding of the designed vaccine to TLR4 receptor. The best structure had RMSD about 23.8 +/- 0.1 with the Z-score of -2.2. The Van der Waals, electrostatic, and de-solvation energy were -46.8 +/- 5.5, -268.1 +/- 28.8, and -3.5 +/- 3.8 kcal.mol-1, respectively. Then, contributing amino acid residues were determined in designed vaccine and TLR4 receptor; residues 3-6, 29-32, 84-105 and 115-124 from the designed vaccine, residues 528-532, 552-557, 577-584 and 604-623 from chain B of TLR4 receptor, and residues 80-88 and 128-132 from chain D of TLR4 (Figure 4).
In the present study, HADDOCK server was used to predict the binding of the designed vaccine to TLR4 receptor. The best structure had RMSD about 23.8 +/- 0.1 with the Z-score of -2.2. The Van der Waals, electrostatic, and de-solvation energy were -46.8 +/- 5.5, -268.1 +/- 28.8, and -3.5 +/- 3.8 kcal.mol-1, respectively. Then, contributing amino acid residues were determined in designed vaccine and TLR4 receptor; residues 3-6, 29-32, 84-105 and 115-124 from the designed vaccine, residues 528-532, 552-557, 577-584 and 604-623 from chain B of TLR4 receptor, and residues 80-88 and 128-132 from chain D of TLR4 (Figure 4).
Fig. 4. Molecular docking of designed vaccine with TLR4 (PDB ID- 4G8A). (A) Docked adjuvant-TLR4 complex (wheat and red) with designed vaccine (blue and green). (B) Interface active residues for designed vaccine (green) with adjuvant-TLR4 (red). The spatial position of the ligand binding to receptor is shown from four angles.
Elastic-mode analysis of the vaccine-TLR4 complex:
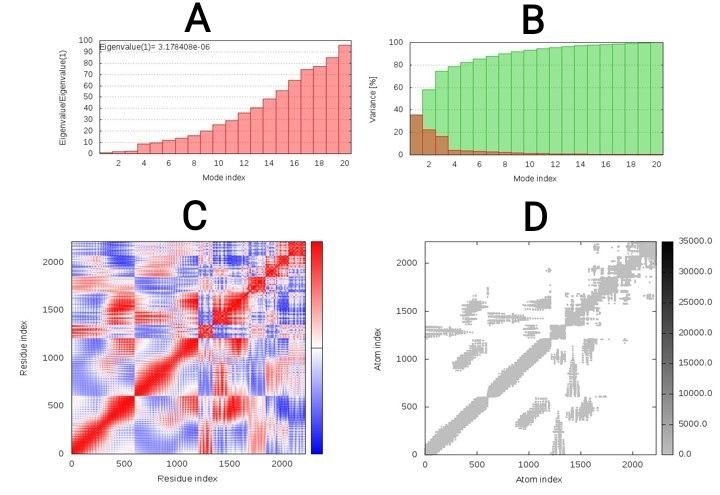
Considering the internal coordinates of the docked complex, normal mode analysis (NMA) was conducted to predict protein-protein interaction stabilization. Individual distortion of residues can impact the complex's deformability using chain hinges. After inverting the variance correlated with each normal mode, the eigenvalue was obtained 3.178408e-08. Using normal mode analysis, B-factor values were obtained which were proportional to RMS. Red, blue, and white colors in the covariance matrix revealed the various pairs of related, anti-correlated or uncorrelated residues motions, respectively. To distinguish the pairs of atoms linked through springs, elastic model of the network was predicted. Each spring was shown by a dot in the diagram, so that colored by the degree of stiffness, between the corresponding atom pairs. Darker greys revealed the more rigid springs (Figure 5).
Fig. 5. Elastic-mode analysis of the vaccine-TLR4 complex, showing (A) eigenvalue; (B) variance %; (C) covariance matrix; (D) elastic network analysis.
Molecular dynamic simulation (MDS):
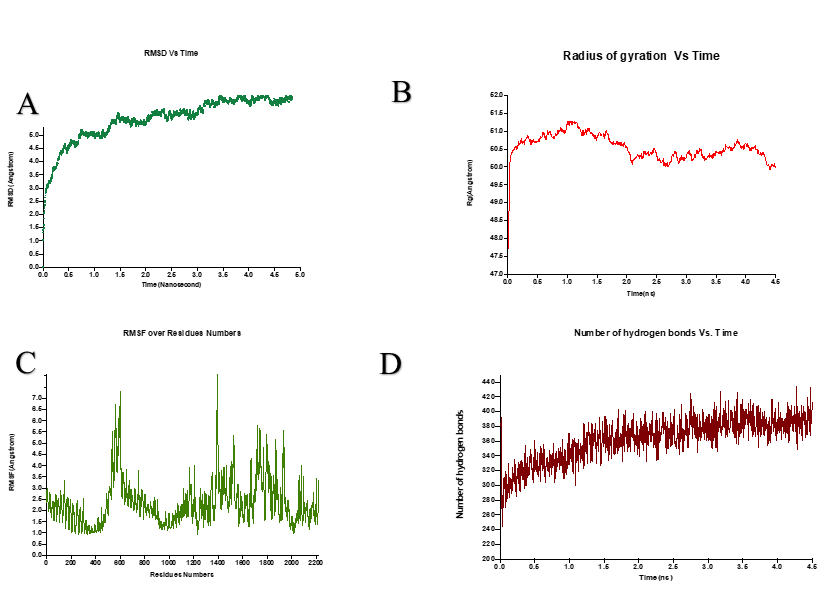
As shown in figure 6 A, the highest peak and average of root square deviation (RMSD) plot of TLR4-4 were 8.1 Å and 6.8 Å, respectively. The RMSD is a parameter of variation of protein that can be used to determine protein stability. Obviously, this construct showed significant changes at initial nanoseconds, but during the rest of this simulation, the plot shows the vaccine-TLR4 complex relatively stabilized after about two nanoseconds with a standard deviation of 0.8 and minor variation over time of the simulation, and it seems to indicate stability in this docked complex.
The plot of the radius of gyration (Rg) indicated the vaccine-TLR4 complex structure compactness (Figure 6 B). A more significant Rg plot variation points to less compactness and vice versa. The average radius of gyration (Rg) in this system was estimated to be 50.5 Å (maximum= 51.2 and minimum- 47.7), and its standard deviation was 0.035. Besides, during the MD simulation time, the Rg plot of this complex showed decreasing trend somewhat, indicating that this protein complex structure attained a more compact and stable structure. The Rq value of this system may decrease with increasing simulation time, and as a result, this structure becomes more compact.
We also used root mean square fluctuations (RMSF) of C-alpha atoms to figure out how flexible each amino acid residue in the vaccine-TLR4 complex is and provide an overall picture of the protein residue fluctuation. The RMSF values range between 0.93 and 8.6807, with an average RMSF of 2.3 for this system. The peaks fluctuations in this RMSF graph at amino acids number GLY1393, PRO1394, GLY1395, and CYS600, PRO601, and GLN561 with 8.07, 8.05, 7.39, 7.32, 7.05, and 6.7 Å, respectively, indicating the existence of highly flexible areas (Figure 6 C).
The number of hydrogen bonds generated during MD simulation time was also used to measure the strength of intermolecular interactions with VMD. Hydrogen bonds play an essential role in protein structure stabilization. The vaccine-TLR4 complex created an average of 362, showing that the vaccine-TLR4 complex and the TLR4 receptor had good intermolecular interactions (Figure 6 D).
Based on the plot, this graph also increased dramatically during the time. These patterns revealed that the designed vaccine's interactions with the TLR4 improved the number of hydrogen bonds, resulting in a more stable vaccine-TLR4 complex during the MD simulation duration.
As shown in figure 6 A, the highest peak and average of root square deviation (RMSD) plot of TLR4-4 were 8.1 Å and 6.8 Å, respectively. The RMSD is a parameter of variation of protein that can be used to determine protein stability. Obviously, this construct showed significant changes at initial nanoseconds, but during the rest of this simulation, the plot shows the vaccine-TLR4 complex relatively stabilized after about two nanoseconds with a standard deviation of 0.8 and minor variation over time of the simulation, and it seems to indicate stability in this docked complex.
The plot of the radius of gyration (Rg) indicated the vaccine-TLR4 complex structure compactness (Figure 6 B). A more significant Rg plot variation points to less compactness and vice versa. The average radius of gyration (Rg) in this system was estimated to be 50.5 Å (maximum= 51.2 and minimum- 47.7), and its standard deviation was 0.035. Besides, during the MD simulation time, the Rg plot of this complex showed decreasing trend somewhat, indicating that this protein complex structure attained a more compact and stable structure. The Rq value of this system may decrease with increasing simulation time, and as a result, this structure becomes more compact.
We also used root mean square fluctuations (RMSF) of C-alpha atoms to figure out how flexible each amino acid residue in the vaccine-TLR4 complex is and provide an overall picture of the protein residue fluctuation. The RMSF values range between 0.93 and 8.6807, with an average RMSF of 2.3 for this system. The peaks fluctuations in this RMSF graph at amino acids number GLY1393, PRO1394, GLY1395, and CYS600, PRO601, and GLN561 with 8.07, 8.05, 7.39, 7.32, 7.05, and 6.7 Å, respectively, indicating the existence of highly flexible areas (Figure 6 C).
The number of hydrogen bonds generated during MD simulation time was also used to measure the strength of intermolecular interactions with VMD. Hydrogen bonds play an essential role in protein structure stabilization. The vaccine-TLR4 complex created an average of 362, showing that the vaccine-TLR4 complex and the TLR4 receptor had good intermolecular interactions (Figure 6 D).
Based on the plot, this graph also increased dramatically during the time. These patterns revealed that the designed vaccine's interactions with the TLR4 improved the number of hydrogen bonds, resulting in a more stable vaccine-TLR4 complex during the MD simulation duration.
mRNA secondary structure prediction:
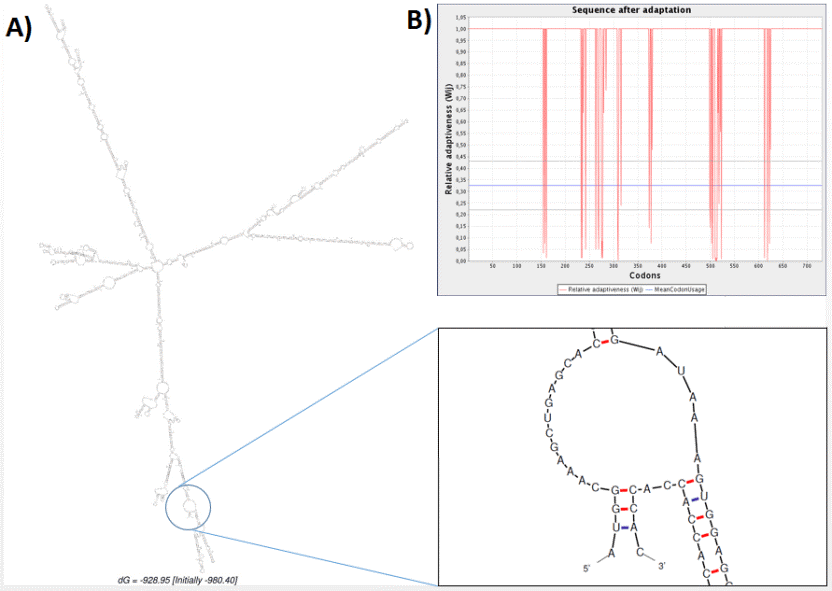
mRNA secondary structure-related free energy at the 5′ end was predicted. The ss value and minimum energy was scored 13.49 ± 13.72 and -980.40 kcal.mol-1, respectively. Neither hairpin nor pseudoknot was observed at 5′ side as shown in figure 7. Moreover, point energy diagram shows two stability indexes. Lower triangle shows optimal energy status and upper triangle plot base pairs. Every colored dot represents a specified energy rate (Figure 8A). Using the circle graph, mRNA structure base pairs status was shown appropriately and the angles were provided to draw un-overlapped helices structure. Red line shows G-C arcs. Blue dashes represent A-U, A-T arcs. G-U, G-T arcs are drawn in green and other arcs are drawn in yellow (Figure 7B). These indices confirmed that the secondary structure of the vaccine mRNA is stable in the bacterial host.
Fig. 7. Analysis of the related RNA structures. (A) Prediction of RNA secondary structure of chimeric gene has no hairpin and
pseudo knot at 5'site of mRNA. (B) Graphical view of codon usage in optimized chimeric gene.
pseudo knot at 5'site of mRNA. (B) Graphical view of codon usage in optimized chimeric gene.
Codon optimization:
The coding sequence of the recombinant vaccine was optimized for expression in E. coli using GenScript’s Multi-parameter Gene Optimization algorithm, OptimumGeneTM (http://www.genescript.com/). The codon optimized sequence had a length of 2205 nucleotides. The codon adaptation index (CAI), the frequency of optimal codons (FOC), and the GC contents of the coding sequence were respectively 0.95, 84 %, and 61% following optimization, indicating the possibility of overexpression of the multi-epitope vaccine in the E. coli host. The suitable GC content is between 30% and 70%. Optimum over-expression of inserted sequence is more than 0.8 according to CAI. Un-adapted codons can be resulted in a minor expression rate. CAI score was also confirmed by JCat. Details of adapted codons are shown in figure 6B. Finally, the coding sequence of designed vaccine was inserted into pET28a (+) and the recombinant plasmid was designed by VectorNTI (Figure 9).
Fig. 8. (A) Point energy diagram for mRNA structure. (B) Circular energy diagram for mRNA structure.
Fig. 9. In silico cloning of the final vaccine construct into pET28a (+) expression vector, where the red part indicates the coding gene for the vaccine surrounded between XhoI (158) and NcoI (2360) while the vector backbone is shown in a black circle.
Discussion
While more than 100 years have passed since the first outbreak of influenza (flu) in 1918, we have faced the emergence of a novel pandemy, causing severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) at the beginning of 2020 (66). What sets this pandemic apart from others is the ability to transmit, which has spread to all continents and affected about 5 million people worldwide in a five-month period (67). The presence of mutations and antigen variability in the coronavirus structure has made it challenging to design and produce a specific vaccine (68).
In the present study, we designed a new chimeric protein vaccine consisting of 27 immunodominant epitopes from the spike protein of SARS-CoV-2. Spike glycoprotein, as a surface antigen with a crown-like appearance, is responsible for the initial binding of the coronavirus to lung cells. Therefore, it plays a pivotal role in entering the host cells (69). SARS-COV-2 spike protein has been shown to be phylogenetically close to bat coronavirus and binds with high affinity to ACE2-derived protein of human origin (70). Studies have shown that mice vaccinated with SARS-like coronaviruses S (SL-CoV) recombinant protein (which are structurally very similar to spike protein of SARS‐CoV‐2) have mounted strong humoral and cellular immune responses by producing high levels of IFN-γ and IL-6. Therefore, spike protein can be considered a suitable target to design vaccines against SARS-CoV-2 (71, 72).
Before the appearance of COVID-19, in 2008, the spike protein-related DNA vaccine was also used to evaluate anti-SARS immunity. In the study by Julie E et al., the vaccine was well tolerated. ELISA detected SARS-CoV-specific antibodies in 80% of cases, and neutralizing antibodies were detected in all individuals. SARS-CoV-specific T4 CD4 + SAR cellular responses were recognized in all cases and T + CD8 cell responses in 20% of individuals (73). Taken together, many vaccine platforms for SARS-CoV have employed spike protein in vaccine structures for immunization, including live attenuated recombinant virus, recombinant modified vaccinia Ankara (MVA) virus, recombinant non-replicating adenovirus (E-deleted), DNA-based vaccines, soluble proteins/adjuvant, virus-like particles (VLPs)/adjuvant, and a combination of vaccine approaches (DNA/peptides, DNA/recombinant viral vector, viral vector/peptides) (74).
In order to boost the immunogenicity of the chimeric vaccine, adjuvant was utilized. Meanwhile, the adjuvant joined with a vaccine intensifies its immunogenic potential and reduces the optimum dose. By mixing with a soluble and weak immunogen, an adjuvant can turn it into an insoluble and stronger immunogen because it retains its high concentration for a longer period of time using antigen depot. In fact, adjuvants are non-specific stimuli for dendritic cells-related to draining as well as cellular and humoral immune responses (75). Recently, TLR agonists have been used as powerful natural adjuvants in vaccine production. Adjuvants enhance the production of inflammatory cytokines, resulting in recruitment of cells of innate and adaptive immunity. In addition to the properties, it strengthens the intracellular cascades that produce inflammatory cytokines and further recruitment of the innate and adaptive immune system cells by stimulate the TLRs of antigen-presenting cells (APC) (76). In this study, the 50S ribosomal protein L7/L12 of Mycobacterium tuberculosis was utilized as an adjuvant and TLR4 agonist. by interacting with TLR4, the 50s ribosomal L7/L12 stimulates the proliferation and activation of immature dendritic and naïve T cells (58, 77).
Accordingly, we selected our favorite sequences considering T and B cell stimulation, high-affinity MHC Class I, MHC Class II, and linear B-cell epitopes from SARS‐CoV‐2 spike protein along with 50s ribosomal L7 / L12. The selected sequences were joined together via appropriate linkers to produce a stable multi-epitope peptide construct. Linkers play a key role in the area of recombinant fusion protein technology such as constructing multi-epitope vaccines. They help the proper folding of each functional domain within the structure of the multi-domain protein, increase the yield of expression in the host systems, and enhance the bioactivity of the recombinant fusion protein (78). In the present study, three types of linkers were used for vaccine construction. The AAY and GPGPG between the epitopes give the structure the ability to minimize junctional immunogenicity (79). AAY is a cleavage site of proteasomes thereby improving the epitope presentation by APCs to immune cells (80). Besides, GPGPG can facilitate the immune processing and epitope presentation by HLA-II to helper T lymphocytes, and is an effective stimulus for cellular immunity, as well (81). EAAAK linker was also used to bind viral antigens to the adjuvants. This linker improves the biological activity of epitopes and intensifies the expression of the final product in the bacterial host, creating a coherent structure (82). The absence of the linkers between the epitopes leads to the production of connective epitopes (neo-epitopes) and protein function disturbance (83).
Therefore, our multi-subunit vaccine sequence was selected with the 1029 length residues. The vaccine had interferon-γ (IFN γ) epitopes. The importance of IFN γ epitopes analysis is due to the fact that IFN-γ produced by CD4+ T cells improve neutrophils and macrophages recruitment using CXC family chemokines and functionally intensify the HTLs and CTLs (84). The presence of 149 positive IFN-γ inducing peptides within the structure of the multi-epitope vaccine implies that the vaccine can enhance the responses of cellular immunity vaccines carrying multiple epitopes which often have poor immunogenicity and require binding to amplifier compounds (85).
The antigenicity score was 0.4606 (close to threshold: 0.4) for the S protein-based vaccine. However, this score increased to 0.835593 when the sequence of the adjuvant was added to the vaccine. When designing a vaccine, it is also crucial to prevent or at least minimize the allergic reactions i.e. the vaccine should prevent the shift in Th1/Th2 balance towards Th2-type response (86). The molecular weight of the vaccine candidate was 76863.88 Da, so overexpressed recombinant protein solubility is expected to be acceptable in E. coli host. The isoelectric pH and the net charge of the protein were 8.66 and +7, respectively, indicating the slightest basic molecule in nature. The instability index was calculated 20.46, indicating that the vaccine is stable upon expression. Moreover, the aliphatic index was 76.18 indicating that there are appropriate aliphatic side chains which make the protein potentially thermostable. Furthermore, grand average of hydropathicity (GRAVY) of -0.164 represents hydrophilicity and efficient interaction with water molecules for better vaccine solubility (87). The solubility of the designed vaccine was also predicted by the SOLpro server.
Understanding the conformational structure of a target protein and their receptors is valuable, where it is possible to determine protein-protein interactions stability and eliminate structural disorders (88). Five 3D structures were predicted for the designed multi-epitope vaccine among which one model with high accuracy and high C-score (-1.15) was selected, and the structural similarities of the vaccine with native protein structures was investigated by considering the TM-score and RMSD. TM-score of 0.57 ± 0.15 and an RMSD of 10.9 ± 4.6% showed that the construct had a correct topology. The selected model was then refined 27% using Galaxy Refine server.
The Ramachandran plot showed whether the residues were found in the favored and allowed regions to confirm the refinement. Comparing the phi and psi dihedral angles of amino acid residues, Ramachandran plot disclosed that there are very few residues in the outlier and unfavorable regions. The ProSA-web server also showed that the original model has been modified (Z-Score=-3.15 to -3.55) after refinement and the designed structure is functionally approached by native proteins. Evaluation of the final three-dimensional structure in terms of discontinuous B-cell epitopes, from a practical point of view showed that epitopes 4 to 115 in length residues could interestingly stimulate antibody production. Immunogenic epitopes exposed on the vaccine structure can effectively induce the differentiation of the B cells to memory and plasma cells, resulting in more persistent protection against SARS-CoV-2 (89). Choudhury et al. reported that TLR4 plays a central role in identification of the molecular patterns of SARS-CoV-2 to generating inflammatory responses. TLR4 can interact with SARS-CoV-2 spike glycoprotein with a higher affinity than TLR1 and TLR6 (90). In vivo studies have also shown that TLR4 knockout mice have either similar survival rates in some cases or even more severe infection than wild-type mice infected with dengue virus (DENV), Ebola virus (EBOV), SARS-CoV, or respiratory syncytial virus (RSV) (91-94). Overall, it was found that virus infection-associated immune response may require TLR4 signaling activation (95).
Therefore, investigating the potential binding strength of the vaccine to TLR4 can be considered as a critical factor in evaluating its efficacy. Hence, we selected the best possible interaction model considering the docking process between the designed construct and TLR4, and evaluated the stability of the complex using elastic-mode analysis. The results showed that the TLR4: vaccine complex had admissible stability, so there is enough time to convey downstream signaling pathways.
The flexibility, compactness, and stability of purposed vaccine structure were also estimated using NAMD and VMD. The complex had a tendency to stabilize after about two nanoseconds and attained a more compact and stable structure during the MD simulation time. This structure may become more compact if the simulation time are increased. Overall picture of the protein residue fluctuation showed the existence of highly flexible areas and the strength of intermolecular interactions with VMD improved the number of hydrogen bonds, resulting in a more stability.
Rigid body docking, clustering of lowest energy structure, and structural refinement were consecutively conducted using energy minimization. The complex with the lowest energy was selected by the server to minimize the potential energy and to obtain stabilized docked complex. Replacing the appropriate protein atoms, inappropriate conformational structure was stabled using energy minimization. Moreover, construct motion stiffness was evaluated by eigenvalue to refine complex deformability. The relative stiffness was appropriately obtained as a functional 3D protein structure. The flexibility of a functional protein structure must be such that its conformational epitopes do not alter in different presumed physiological conditions. Ultimately, the production potential must be evaluated in a bacterial expression host to investigate the effectiveness of a designed vaccine. From a practical point of view, the gene sequence of the vaccine of the interest must be stable enough to be translated following the transcription process (96). Hairpin and pseudoknot at the 5' end of the secondary structure of mRNA can adversely affect the efficiency of the protein translation. The sequence length, the number and situation of loops, the GC content, and etc. must be evaluated before the in-vitro phase. Examination of these important indicators showed the stable status of the mRNA structure (97). The protein can be purified to assess the quality of its binding to the antibodies present in the serum of SARS- CoV-19 infected patient using serological tests. Therefore, codons were optimized in order to over-express the vaccine protein in a suitable host (E. coli strain K12). CAI, FOC, and GC Content were reported 0.95, 84%, and 61%, respectively, demonstrating the desirable overexpression potential. Changes in mentioned parameters can also have profound effects on gene expression. Based on the finding, it was predictable that inserting the vaccine sequence in the vector pET28a (+) would have an acceptable expression in E. coli.
Conclusion
Nowadays, a worldwide effort is being made to prevent and treat emerging viral infections. Meanwhile, predicting the efficiency of the vaccines before their production has become the main approach of all studies. That is why in silico studies have become an intelligent strategy for producing vaccines. Accordingly, in the present study, a multi-subunit vaccine was designed against SARS-CoV-2 using the antigen minimization approach. The vaccine was designed based on the most conserve and immunogenic epitopes of the SARS-CoV-2 spike protein, instead of focusing on the whole virus. The scores obtained from the immunoinformatics databases indicated that the construct could be considered an appropriate vaccine candidate to protect against COVID-19 infection. Indeed, in vitro and in vivo studies need to be performed to evaluate the production of the vaccine in prokaryotic host systems, as well as, investigating its efficacy in eliciting the immune responses in animal models.
Acknowledgments
None declared.
Conflict of interest
The authors have no conflict of interest in this study.
Funding/support
This study was financially supported by the office of vice-chancellor for research of Shiraz University of Medical Sciences.
Data availability
The raw data supporting the conclusions of this article are available from the authors upon reasonable request.
Type of Study: Research Article |
Subject:
Advanced Sciences and Technologies in Public Health
Received: 2023/05/3 | Accepted: 2023/06/21 | Published: 2023/07/19
Received: 2023/05/3 | Accepted: 2023/06/21 | Published: 2023/07/19
References
1. Wang N, Shang J, Jiang S, Du L. Subunit vaccines against emerging pathogenic human coronaviruses. Frontiers in Microbiology. 2020;11:298. [DOI] [PMID] [PMCID]
2. King AM, Lefkowitz EJ, Mushegian AR, Adams MJ, Dutilh BE, Gorbalenya AE, et al. Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2018). Archives of virology. 2018;163(9):2601-31. [DOI] [PMID]
3. Khatami SH, Movahedpour A, Taheri-Anganeh M. Serological and RT-PCR diagnosis approaches for COVID-19. Health Science Monitor. 2022;1(1):24-31. [DOI]
4. Drosten C, Günther S, Preiser W, Van Der Werf S, Brodt H-R, Becker S, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. New England journal of medicine. 2003;348(20):1967-76.
https://doi.org/10.1056/NEJMoa030747 [DOI] [PMID]
5. Azhar EI, Hui DS, Memish ZA, Drosten C, Zumla A. The Middle East Respiratory Syndrome (MERS). Infectious Disease Clinics. 2019;33(4):891-905. [DOI] [PMID] [PMCID]
6. Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. New England Journal of Medicine. 2020. [Google Scholar]
7. Prompetchara E, Ketloy C, Palaga T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pac J Allergy Immunol. 2020;38(1):1-9.
https://doi.org/10.12932/AP-200220-0772 [DOI]
8. Ghelichi-Ghojogh M, Khezri R, Rezaei F, Aljalili S, Valizadeh R, Sadighpour T. Health inequality in COVID-19 vaccination coverage. Health Science Monitor. 2023;2(1):10-2. [DOI]
9. Wu F, Zhao S, Yu B, Chen Y-M, Wang W, Song Z-G, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265-9. [DOI] [PMID] [PMCID]
10. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet. 2020;395(10223):497-506.
https://doi.org/10.1016/S0140-6736(20)30183-5 [DOI] [PMID]
11. Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. The lancet. 2020;395(10224):565-74.
https://doi.org/10.1016/S0140-6736(20)30251-8 [DOI] [PMID]
12. Han Q, Lin Q, Jin S, You L. Coronavirus 2019-nCoV: A brief perspective from the front line. Journal of Infection. 2020;80(4):373-7. [DOI] [PMID] [PMCID]
13. Ahmed SF, Quadeer AA, McKay MR. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses. 2020;12(3):254. [DOI] [PMID] [PMCID]
14. Rahman MS, Hoque MN, Islam MR, Akter S, Rubayet-Ul-Alam A, Siddique MA, et al. Epitope-based chimeric peptide vaccine design against S, M and E proteins of SARS-CoV-2 etiologic agent of global pandemic COVID-19: an in silico approach. BioRxiv. 2020. [DOI]
15. Srivastava S, Verma S, Kamthania M, Kaur R, Badyal RK, Saxena AK, et al. Structural basis to design multi-epitope vaccines against Novel Coronavirus 19 (COVID19) infection, the ongoing pandemic emergency: an in silico approach. bioRxiv. 2020. [DOI]
16. Hoffmann M, Kleine-Weber H, Krüger N, Mueller MA, Drosten C, Pöhlmann S. The novel coronavirus 2019 (2019-nCoV) uses the SARS-coronavirus receptor ACE2 and the cellular protease TMPRSS2 for entry into target cells. BioRxiv. 2020. [DOI]
17. Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. Journal of medical virology. 2020;92(4):418-23. [DOI] [PMID] [PMCID]
18. Nourani A, Rahimkhoei V, Akbari A. Potential therapeutic drug candidates against SARS-CoV-2 (COVID‐19) through molecular docking: A review. Health Science Monitor. 2022;1(2):89-106. [DOI]
19. Chan JF-W, Kok K-H, Zhu Z, Chu H, To KK-W, Yuan S, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerging Microbes & Infections. 2020;9(1):221-36. [DOI] [PMID] [PMCID]
20. Li F. Structure, function, and evolution of coronavirus spike proteins. Annual review of virology. 2016;3:237-61. [DOI] [PMID] [PMCID]
21. Song W, Gui M, Wang X, Xiang Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS pathogens. 2018;14(8):e1007236. [DOI] [PMID] [PMCID]
22. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260-3. [DOI] [PMID] [PMCID]
23. Saha R, Prasad BV. In silico approach for designing of a multi-epitope based vaccine against novel Coronavirus (SARS-COV-2). bioRxiv. 2020. [DOI]
24. Behbahani M. In silico Design of novel Multi-epitope recombinant Vaccine based on Coronavirus surface glycoprotein. bioRxiv. 2020. [DOI]
25. Commission GOoNH. General Office of National Administration of Traditional Chinese Medicine. Diagnostic and treatment protocol for Novel Coronavirus Pneumonia. 2020. [DOI]
26. Jin Y, Yang H, Ji W, Wu W, Chen S, Zhang W, et al. Virology, Epidemiology, Pathogenesis, and Control of COVID-19. Viruses. 2020;12(4):372. [DOI] [PMID] [PMCID]
27. He X-J, Chen S-Y, Wu J-P, Yang L-R, Xu G. Highly efficient enzymatic synthesis of tert-butyl (S)-6-chloro-5-hydroxy-3-oxohexanoate with a mutant alcohol dehydrogenase of Lactobacillus kefir. Applied microbiology and biotechnology. 2015;99(21):8963-75. [DOI] [PMID]
28. Nosrati M, Mohabatkar H, Behbahani M. A novel multi-epitope vaccine for cross protection against hepatitis C virus (HCV): an immunoinformatics approach. Research in Molecular Medicine. 2017;5(1):17-26. [DOI]
29. Jain N, Shankar U, Majee P, Kumar A. Scrutinizing the SARS-CoV-2 protein information for the designing an effective vaccine encompassing both the T-cell and B-cell epitopes. BioRxiv. 2020. [DOI]
30. Yousefi T, Mir SM, Asadi J, Tourani M, Karimian A, Maniati M, et al. In silico analysis of non-synonymous single nucleotide polymorphism in a human KLK-2 gene associated with prostate cancer. Meta Gene. 2019;21:100578. [DOI]
31. Taheri-Anganeh M, Khatami SH, Jamali Z, Savardashtaki A, Ghasemi Y, Mostafavi-Pour Z. In silico analysis of suitable signal peptides for secretion of a recombinant alcohol dehydrogenase with a key role in atorvastatin enzymatic synthesis. Molecular biology research communications. 2019;8(1):17-26. [DOI]
32. Taheri-Anganeh M, Amiri A, Movahedpour A, Khatami SH, Ghasemi Y, Savardashtaki A, et al. In silico Evaluation of PLAC1-fliC As a Chimeric Vaccine against Breast Cancer. Iranian Biomedical Journal. 2020;24(3):173-82. [DOI] [PMID] [PMCID]
33. Vafadar A, Taheri-Anganeh M, Movahedpour A, Jamali Z, Irajie C, Ghasemi Y, et al. In Silico Design and Evaluation of scFv-CdtB as a Novel Immunotoxin for Breast Cancer Treatment. Int J Cancer Manag. 2020;13(1):e96094. [DOI]
34. Khatami SH, Taheri-Anganeh M, Arianfar F, Savardashtaki A, Sarkari B, Ghasemi Y, et al. Analyzing Signal Peptides for Secretory Production of Recombinant Diagnostic Antigen B8/1 from Echinococcus granulosus: An In silico Approach. Molecular Biology Research Communications. 2020:1-10. [DOI]
35. Asadi M, Gharibi S, Khatami SH, Shabaninejad Z, Kargar F, Yousefi F, et al. Analysis of Suitable Signal Peptides for Designing a Secretory Thermostable Cyanide Degrading Nitrilase: An in Silico Approach. Journal of Environmental Treatment Techniques. 2019;7(3):506-13. [Google Scholar]
36. Abdelmageed MI, Abdelmoneim AH, Mustafa MI, Elfadol NM, Murshed NS, Shantier SW, et al. Design of multi epitope-based peptide vaccine against E protein of human 2019-nCoV: An immunoinformatics approach. BioRxiv. 2020. [DOI]
37. Shahid F, Ashfaq UA, Javaid A, Khalid H. Immunoinformatics guided rational design of a next generation multi epitope based peptide (MEBP) vaccine by exploring Zika virus proteome. Infection, Genetics and Evolution. 2020;80:104199. [DOI] [PMID]
38. Ikram A, Zaheer T, Awan FM, Obaid A, Naz A, Hanif R, et al. Exploring NS3/4A, NS5A and NS5B proteins to design conserved subunit multi-epitope vaccine against HCV utilizing immunoinformatics approaches. Scientific reports. 2018;8(1):1-14. [DOI] [PMID] [PMCID]
39. Hajighahramani N, Eslami M, Negahdaripour M, Ghoshoon MB, Dehshahri A, Erfani N, et al. Computational design of a chimeric epitope-based vaccine to protect against Staphylococcus aureus infections. Molecular and cellular probes. 2019;46:101414. [DOI] [PMID]
40. Hajighahramani N, Nezafat N, Eslami M, Negahdaripour M, Rahmatabadi SS, Ghasemi Y. Immunoinformatics analysis and in silico designing of a novel multi-epitope peptide vaccine against Staphylococcus aureus. Infection, Genetics and Evolution. 2017;48:83-94. [DOI] [PMID]
41. Negahdaripour M, Nezafat N, Eslami M, Ghoshoon MB, Shoolian E, Najafipour S, et al. Structural vaccinology considerations for in silico designing of a multi-epitope vaccine. Infection, Genetics and Evolution. 2018;58:96-109. [DOI] [PMID]
42. Farhadi T, Nezafat N, Ghasemi Y, Karimi Z, Hemmati S, Erfani N. Designing of complex multi-epitope peptide vaccine based on omps of Klebsiella pneumoniae: an in silico approach. International Journal of Peptide Research and Therapeutics. 2015;21(3):325-41. [DOI]
43. Purcell AW, McCluskey J, Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nature reviews Drug discovery. 2007;6(5):404-14. [DOI] [PMID]
44. Enjuanes L, Zuniga S, Castano-Rodriguez C, Gutierrez-Alvarez J, Canton J, Sola I. Molecular Basis of Coronavirus Virulence and Vaccine Development. Advances in virus research. 2016;96:245-86. [DOI] [PMID] [PMCID]
45. Baruah V, Bose S. Immunoinformatics-aided identification of T cell and B cell epitopes in the surface glycoprotein of 2019-nCoV. J Med Virol. 2020;92(5):495-500. [DOI] [PMID] [PMCID]
46. Jespersen MC, Peters B, Nielsen M, Marcatili P. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic acids research. 2017;45(W1):W24-W9. [DOI] [PMID] [PMCID]
47. Saha S, Raghava GPS. Prediction of continuous B‐cell epitopes in an antigen using recurrent neural network. Proteins: Structure, Function, and Bioinformatics. 2006;65(1):40-8. [DOI] [PMID]
48. Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC bioinformatics. 2007;8(1):424. [DOI] [PMID] [PMCID]
49. Bhasin M, Raghava G. Prediction of CTL epitopes using QM, SVM and ANN techniques. Vaccine. 2004;22(23-24):3195-204. [DOI] [PMID]
50. Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, Yan Z, et al. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology. 2018;154(3):394-406. [DOI] [PMID] [PMCID]
51. Magnan CN, Zeller M, Kayala MA, Vigil A, Randall A, Felgner PL, et al. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics. 2010;26(23):2936-43. [DOI] [PMID] [PMCID]
52. Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC bioinformatics. 2007;8(1):4. [DOI] [PMID] [PMCID]
53. Dimitrov I, Bangov I, Flower DR, Doytchinova I. AllerTOP v. 2-a server for in silico prediction of allergens. Journal of molecular modeling. 2014;20(6):2278. [DOI] [PMID]
54. Dimitrov I, Naneva L, Doytchinova I, Bangov I. AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics. 2014;30(6):846-51. [DOI] [PMID]
55. Gasteiger E, Hoogland C, Gattiker A, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. The proteomics protocols handbook: Springer; 2005. p. 571-607. [DOI]
56. Magnan CN, Randall A, Baldi P. SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics. 2009;25(17):2200-7. [DOI] [PMID]
57. Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nature protocols. 2010;5(4):725. [DOI] [PMID] [PMCID]
58. Shey RA, Ghogomu SM, Esoh KK, Nebangwa ND, Shintouo CM, Nongley NF, et al. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Scientific reports. 2019;9(1):1-18. [DOI] [PMID] [PMCID]
59. Heo L, Park H, Seok C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic acids research. 2013;41(W1):W384-W8. [DOI] [PMID] [PMCID]
60. Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic acids research. 2007;35(suppl_2):W407-W10. [DOI] [PMID] [PMCID]
61. Lovell SC, Davis IW, Arendall III WB, De Bakker PI, Word JM, Prisant MG, et al. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins: Structure, Function, and Bioinformatics. 2003;50(3):437-50. [DOI] [PMID]
62. Van Regenmortel MH. Mapping epitope structure and activity: from one-dimensional prediction to four-dimensional description of antigenic specificity. Methods. 1996;9(3):465-72. [DOI] [PMID]
63. Ponomarenko J, Bui H-H, Li W, Fusseder N, Bourne PE, Sette A, et al. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC bioinformatics. 2008;9(1):514. [DOI] [PMID] [PMCID]
64. López-Blanco JR, Aliaga JI, Quintana-Ortí ES, Chacón P. iMODS: internal coordinates normal mode analysis server. Nucleic acids research. 2014;42(W1):W271-W6. [DOI] [PMID] [PMCID]
65. Mauro VP. Codon Optimization in the Production of Recombinant Biotherapeutics: Potential Risks and Considerations. BioDrugs. 2018;32(1):69-81. [DOI] [PMID]
66. Shi Y, Wang Y, Shao C, Huang J, Gan J, Huang X, et al. COVID-19 infection: the perspectives on immune responses. Nature Publishing Group; 2020. [DOI] [PMID] [PMCID]
67. Zheng J. SARS-CoV-2: an emerging coronavirus that causes a global threat. International journal of biological sciences. 2020;16(10):1678. [DOI] [PMID] [PMCID]
68. Chatterjee S. Understanding the nature of variations in structural sequences coding for coronavirus spike, envelope, membrane and nucleocapsid proteins of sars-cov-2. Envelope, Membrane and Nucleocapsid Proteins of SARS-CoV-2 (March 28, 2020). 2020. [DOI]
69. Tehrani SS, Goodarzi G, Naghizadeh M, Khatami SH, Movahedpour A, Abbasi A, et al. In Silico Evaluation of Suitable Signal Peptides for Secretory Production of Recombinant Granulocyte Colony Stimulating Factor in Escherichia coli. Recent Patents on Biotechnology. 2020. [DOI] [PMID]
70. Ortega JT, Serrano ML, Pujol FH, Rangel HR. Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: An in silico analysis. EXCLI journal. 2020;19:410. [DOI] [PMID] [PMCID]
71. Hou Y-x, Peng C, Han Z-g, Zhou P, Chen J-g, Shi Z-l. Immunogenicity of the spike glycoprotein of Bat SARS-like coronavirus. Virologica Sinica. 2010;25(1):36-44. [DOI] [PMID] [PMCID]
72. Choudhury A, Mukherjee S. In silico studies on the comparative characterization of the interactions of SARS‐CoV‐2 spike glycoprotein with ACE‐2 receptor homologs and human TLRs. Journal of Medical Virology. 2020. [DOI] [PMID] [PMCID]
73. Martin JE, Louder MK, Holman LA, Gordon IJ, Enama ME, Larkin BD, et al. A SARS DNA vaccine induces neutralizing antibody and cellular immune responses in healthy adults in a Phase I clinical trial. Vaccine. 2008;26(50):6338-43. [DOI] [PMID] [PMCID]
74. Padron-Regalado E. Vaccines for SARS-CoV-2: lessons from other coronavirus strains. Infectious diseases and therapy. 2020:1-20. [DOI] [PMID] [PMCID]
75. Tourani M, Karkhah A, Najafi A. Development of an epitope-based vaccine inhibiting immune cells rolling and migration against atherosclerosis using in silico approaches. Computational Biology and Chemistry. 2017;70:156-63. [DOI] [PMID]
76. Rahmani A, Baee M, Rostamtabar M, Karkhah A, Alizadeh S, Tourani M, et al. Development of a conserved chimeric vaccine based on helper T-cell and CTL epitopes for induction of strong immune response against Schistosoma mansoni using immunoinformatics approaches. International journal of biological macromolecules. 2019;141:125-36. [DOI] [PMID]
77. Deng H, Yu S, Guo Y, Gu L, Wang G, Ren Z, et al. Development of a multivalent enterovirus subunit vaccine based on immunoinformatic design principles for the prevention of HFMD. Vaccine. 2020. [DOI] [PMID]
78. Movahedpour A, Mostafavi-Pour Z, Sarkari B, Taheri-Anganeh M, Nezafat N, Savardashtaki A, et al. Designing a Multi-Epitope Antigen for Serodiagnosis of Strongyloides stercoralis Based on L3Nie. 01 and IgG Immunoreactive Epitopes. Avicenna Journal of Medical Biotechnology. 2022;14(2):114-24. [DOI] [PMID] [PMCID]
79. Gutiérrez AH, Loving C, Moise L, Terry FE, Brockmeier SL, Hughes HR, et al. In vivo validation of predicted and conserved T cell epitopes in a swine influenza model. PLoS One. 2016;11(7). [DOI] [PMID] [PMCID]
80. Chaudhuri D, Datta J, Majumder S, Giri K. In silico designing of peptide based vaccine for Hepatitis viruses using reverse vaccinology approach. Infection, Genetics and Evolution. 2020:104388. [DOI] [PMID]
81. Validi M, Karkhah A, Prajapati VK, Nouri HR. Immuno-informatics based approaches to design a novel multi epitope-based vaccine for immune response reinforcement against Leptospirosis. Molecular immunology. 2018;104:128-38. [DOI] [PMID]
82. Partovi Nasr M, Motalebi M, Zamani MR, Jourabchi E. In Silico Analysis and Expression of Osmotin-EAAAK-LTP Fused Protein. Journal of Genetic Resources. 2020;6(1):41-8. []
83. Khan M, Khan S, Ali A, Akbar H, Sayaf AM, Khan A, et al. Immunoinformatics approaches to explore Helicobacter Pylori proteome (Virulence Factors) to design B and T cell multi-epitope subunit vaccine. Scientific Reports. 2019;9(1):13321. [DOI] [PMID] [PMCID]
84. Rostamtabar M, Rahmani A, Baee M, Karkhah A, Prajapati VK, Ebrahimpour S, et al. Development a multi-epitope driven subunit vaccine for immune response reinforcement against Serogroup B of Neisseria meningitidis using comprehensive immunoinformatics approaches. Infection, Genetics and Evolution. 2019;75:103992. [DOI] [PMID]
85. Bazhan SI, Antonets DV, Karpenko LI, Oreshkova SF, Kaplina ON, Starostina EV, et al. In silico designed ebola virus T-cell multi-epitope DNA vaccine constructions are immunogenic in mice. Vaccines. 2019;7(2):34. [DOI] [PMID] [PMCID]
86. Jung S-Y, Kang KW, Lee E-Y, Seo D-W, Kim H-L, Kim H, et al. Heterologous prime-boost vaccination with adenoviral vector and protein nanoparticles induces both Th1 and Th2 responses against Middle East respiratory syndrome coronavirus. Vaccine. 2018;36(24):3468-76. [DOI] [PMID] [PMCID]
87. Dauda A, Saul S, Kolo YS. Biocomputational Analysis of Chlamydia abortus Protein Sequence. 2017. [URL]
88. Rubin S, Qvit N. Cyclic Peptides for Protein− Protein Interaction Targets: Applications to Human Disease. Critical Reviews™ in Eukaryotic Gene Expression. 2016;26(3).
https://doi.org/10.1615/CritRevEukaryotGeneExpr.2016016525 [DOI] [PMID]
89. Akkaya M, Kwak K, Pierce SK. B cell memory: building two walls of protection against pathogens. Nature Reviews Immunology. 2019:1-10. [DOI] [PMID] [PMCID]
90. Choudhury A, Mukherjee S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J Med Virol. 2020. [DOI] [PMID] [PMCID]
91. Younan P, Ramanathan P, Graber J, Gusovsky F, Bukreyev A. The Toll-Like Receptor 4 Antagonist Eritoran Protects Mice from Lethal Filovirus Challenge. mBio. 2017;8(2).
https://doi.org/10.1128/mBio.00226-17 [DOI] [PMID] [PMCID]
92. Modhiran N, Watterson D, Blumenthal A, Baxter AG, Young PR, Stacey KJ. Dengue virus NS1 protein activates immune cells via TLR4 but not TLR2 or TLR6. Immunology and cell biology. 2017;95(5):491-5. [DOI] [PMID]
93. Glasner DR, Ratnasiri K, Puerta-Guardo H, Espinosa DA, Beatty PR, Harris E. Dengue virus NS1 cytokine-independent vascular leak is dependent on endothelial glycocalyx components. PLoS pathogens. 2017;13(11). [DOI] [PMID] [PMCID]
94. Marr N, Turvey SE. Role of human TLR4 in respiratory syncytial virus-induced NF-κB activation, viral entry and replication. Innate immunity. 2012;18(6):856-65. [DOI] [PMID]
95. Olejnik J, Hume AJ, Mühlberger E. Toll-like receptor 4 in acute viral infection: Too much of a good thing. PLoS pathogens. 2018;14(12). [DOI] [PMID] [PMCID]
96. Sahin U, Holtkamp S, Tureci O, Kreiter S. Modification of RNA, Producing an Increased Transcript Stability and Translation Efficiency. Google Patents; 2019. [URL]
97. Bakhshi M, Ebrahimi F, Nazarian S, Zargan J. Computational analysis and gene cloning: design and preparation of a multi subunit vaccine consisting of EspA, Stx2B and Intimin antigens against enterohaemrrhagic Escherichia coli. 2018. [Google Scholar]
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
Contact Information
Address: Urmia University of Medical Sciences, Jahad Ave., Urmia, I.R. Iran.
Post code: 57147-83734. P.O. Box. 1138
Email: healthsciencemonitor gmail.com
gmail.com
hsmumsu.ac.ir
Tel / Fax: +984431937415/+984432240642
